👉 Nesse projeto, desenvolvi uma aplicação web no Streamlit, a qual pode ser acessada clicando aqui, que realiza o alinhamento de sequências de DNA.
👉 O código, em Python, da aplicação web pode ser acessado aqui.
Abaixo encontra-se uma breve explicação sobre a importância e sobre os algoritmos de alinhamento de sequências biológicas.
Uma das práticas mais comuns na bioinformática é o alinhamento de sequências biológicas (DNA, RNA e proteínas).

Por que isso é importante?
O alinhamento busca por regiões com similaridades entre duas ou mais sequências, nos fornecendo informações sobre relações funcionais, estruturais e evolutivas dos e entre os seres vivos.
Várias são as aplicações desse método, dentre elas:
- montagem de genomas
- descoberta de novas drogas
- reconstrução da história evolutiva
- buscas em bancos de dados
- análise de variantes
- dedução de função
- definição de regiões conservadas
O alinhamento de sequências pode ser:
- global ou local
- par a par ou múltiplo
Realizamos o alinhamento global quando o objetivo é alinhar toda a extensão das sequências, ou seja, o alinhamento é realizado de ponta a ponta.
O alinhamento global pode ser par a par, quando alinhamos duas sequências:

ou múltiplo, quando três ou mais sequências são alinhadas:
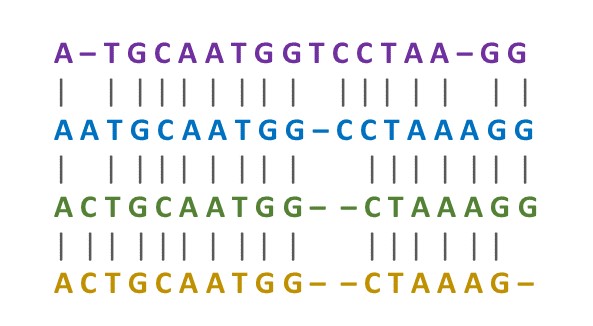
O algoritmo mais utilizado para realizar o alinhamento global par a par é o de Needleman-Wunsch, o qual tem uma resolução ótima. Para o alinhamento múltiplo, CLUSTAL, de Higgins-Sharp, é o algoritmo mais utilizado, com uma resolução aproximada (heurística).
O alinhamento local busca por região de alta similaridade entre subsequências das sequências analisadas, não importando as regiões adjacentes:
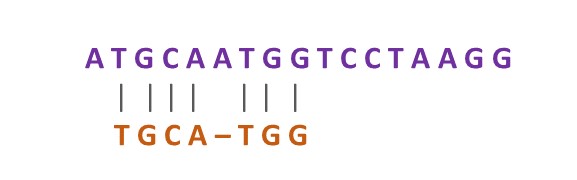
Para isso, o algoritmo mais utilizado é o de Smith-Waterman, que produz uma solução ótima.
Os algoritmos de alinhamento par a par analisam o score de cada alinhamento. O score é o resultado da soma das pontuações para matches (alinhamento de caracteres iguais), mismatches (alinhamento de caracteres diferentes) e gaps (inserção ou deleção de caracteres), sendo que os matches são recompensados, enquanto mismatches e gaps são penalizados.
Vamos analisar como ficaria o alinhamento global entre as duas sequências abaixo:

Primeiro, vamos considerar um alinhamento mais simples, em que mismatches não são permitidos, e que a recompensa para match é +4 e a penalidade para gap é -3:

Por trás dos panos, o algoritmo de Needleman-Wunsch constrói uma matriz pontuando todos os alinhamentos possíveis. Para visualizarmos o alinhamento, fazemos o tracebacking a partir do último valor da matriz e seguimos o caminho do qual cada pontuação se originou, sendo que "↖" indica um match, "↑" um gap na sequência que está na horizontal da matriz, e "←" um gap na sequência que está na vertical da matriz:

Agora vamos analisar um alinhamento que permite mismatches, sendo este com penalidade de -2:

A matriz é semelhante, com a diferença de que agora existe um mismatch entre "A" e "C":
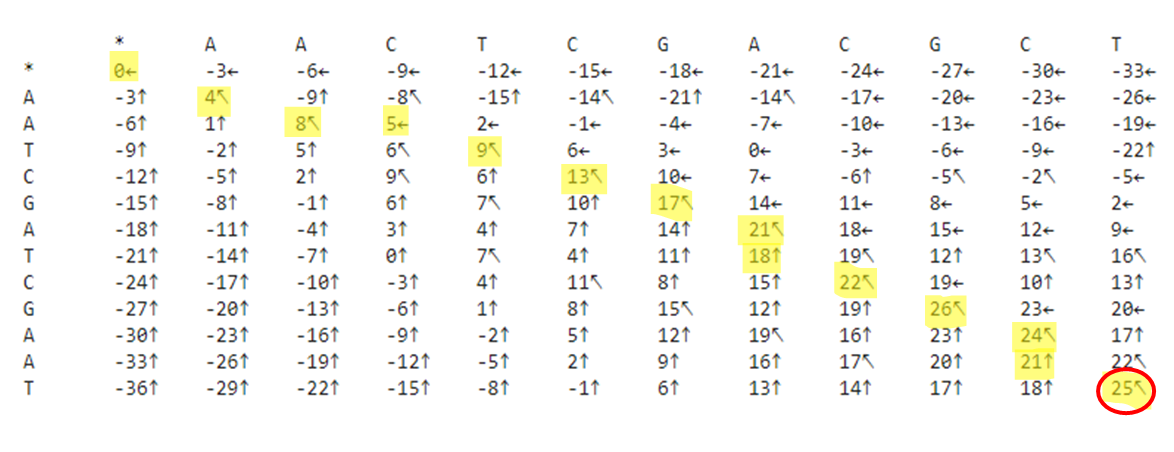
Podemos ver que o maior score está no alinhamento que permite mismatches (score = 25), sendo esse considerado um melhor alinhamento. Isto porque a penalidade para gap é maior visto que, do ponto de vista evolutivo, "há um maior custo" quando acontece uma deleção/inserção de caractere do que quando ocorre uma troca de caractere.
Para o alinhamento local, o algoritmo de Smith-Waterman é uma modificação do de Needleman-Wunsch para que sempre que uma pontuação fica com valor negativo, a matriz é reiniciada com zero. Além disso, o tracebacking não começa pelo último valor, mas sim pelo maior valor da matriz, e termina quando encontra uma pontuação igual a zero. O alinhamento local abaixo teria a seguinte matriz:
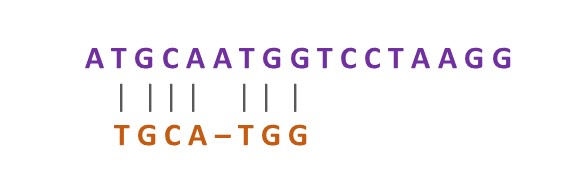
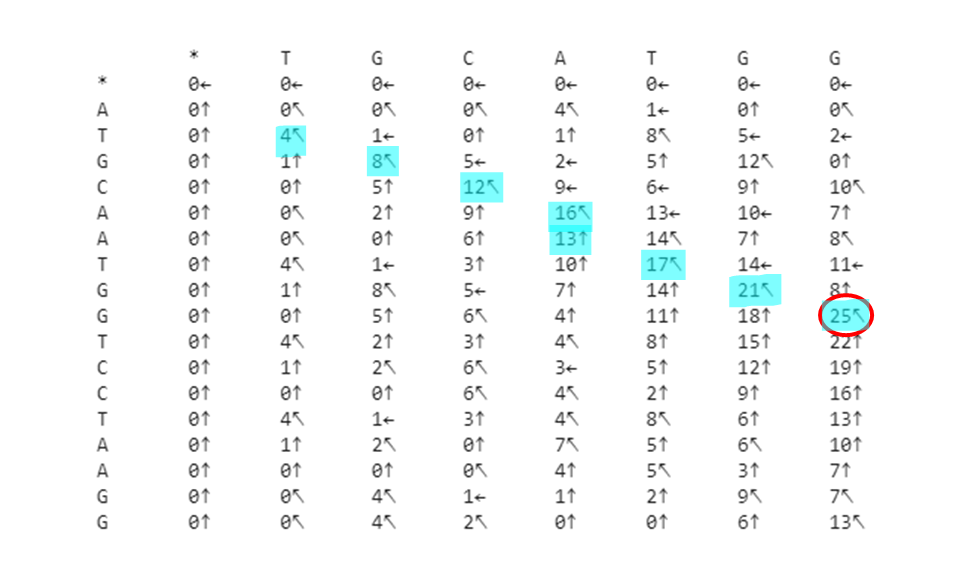
Não existe uma regra que determine qual deve ser a pontuação para match, mismatch e gap em alinhamento de nucleotídeos. Para alinhamento de proteínas, existem as Matrizes de Substituição (PAM, BLOSUM) as quais contêm as probabilidades das trocas ou manutenção dos aminoácidos.
Diante dessas informações, construí uma aplicação web no Streamlit que realiza o alinhamento global e local, par a par, de sequências de DNA, baseado nos algoritmos de Needleman-Wunsch e Smith-Waterman.
Além do alinhamento, esse web app também analisa a Composição de Nucleotídeos e o Conteúdo GC das sequências fornecidas, características importantes em estudos genéticos, evolutivos, taxonômicos e ecológicos, uma vez que fornecem informações sobre o padrão de utilização dos códons, identificação de regiões gênicas, e auxiliam na síntese de vacinas de DNA e no desenho de primers, por exemplo.