- Overview
- Quick Start
- Advanced Configuration
3.1. Optional CloudFormation Parameters
3.2. Manual Data Download - Module Information
- Architecture Details
- Security
- License
Proteins are large biomolecules that play an important role in the body. Knowing the physical structure of proteins is key to understanding their function. However, it can be difficult and expensive to determine the structure of many proteins experimentally. One alternative is to predict these structures using machine learning algorithms. Several high-profile research teams have released such algorithms, including OpenFold, AlphaFold 2, RoseTTAFold ]and others. Their work was important enough for Science magazine to name it the "2021 Breakthrough of the Year".
Many AI-driven folding algorithms use a multi-track transformer architecture trained on known protein templates to predict the structure of unknown peptide sequences. These predictions are heavily GPU-dependent and take anywhere from minutes to days to complete. The input features for these predictions include multiple sequence alignment (MSA) data. MSA algorithms are CPU-dependent and can themselves require several hours of processing time.
Running both the MSA and structure prediction steps in the same computing environment can be cost inefficient, because the expensive GPU resources required for the prediction sit unused while the MSA step runs. Instead, using a high-performance computing (HPC) service like AWS Batch allows us to run each step as a containerized job with the best fit of CPU, memory, and GPU resources.
This repository includes the CloudFormation template, Jupyter Notebook, and supporting code to run protein analysis algorithms on AWS Batch.
-
Choose Launch Stack and (if prompted) log into your AWS account:

-
For Stack Name, enter a value unique to your account and region. Leave the other parameters as their default values and select Next.
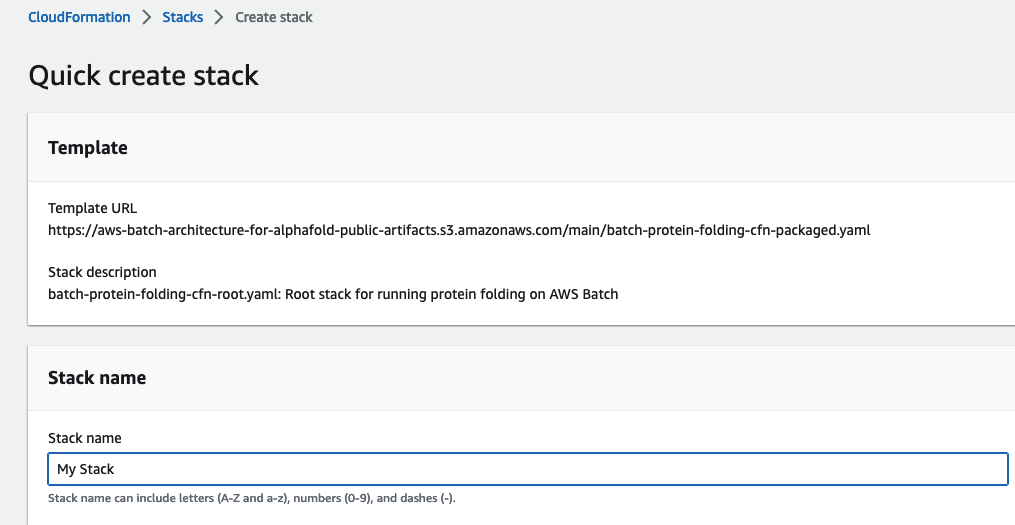
-
Select I acknowledge that AWS CloudFormation might create IAM resources with custom names.
-
Choose Create stack.
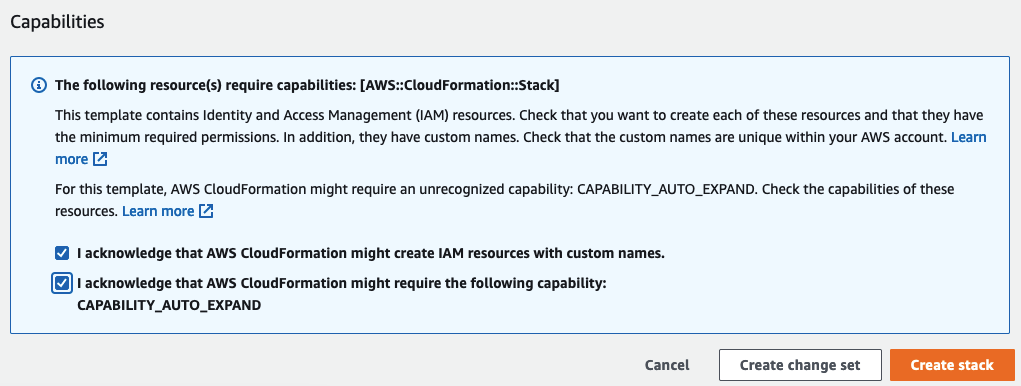
-
Wait 20 minutes for AWS CloudFormation to create the necessary infrastructure stack and module containers.
-
Wait an additional 5 hours for AWS Batch to download the necessary reference data to the Amazon FSx for Lustre file system.
-
Navigate to SageMaker.
-
Select Notebook > Notebook instances.

-
Select the BatchFoldNotebookInstance instance and then Actions > Open JupyterLab.
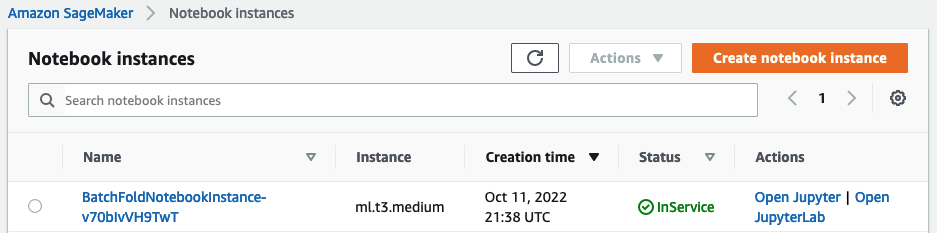
-
Open the quick start notebook at
notebooks/quick-start-protein-folding.ipynb.
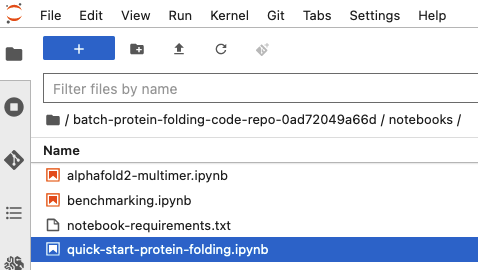
-
Select the conda_python_3 kernel.
-
Run the notebook cells to create and analyze several protein folding jobs.
-
(Optional) To delete all provisioned resources from from your account, navigate to Cloud Formation, select your stack, and then Delete.






- Select "Y" for LaunchSageMakerNotebook if you want to launch a managed sagemaker notebook instance to quickly run the provided Jupyter notebook.
- Provide values for the VPC, Subnet, and DefaultSecurityGroup parameters to use existing network resources. If one or more of those parameters are left empty, CloudFormation will create a new VPC and FSx for Lustre instance for the stack.
- Provide values for the FileSystemId and FileSystemMountName parameters to use an existing FSx for Lustre file system. If one or more of these parameters are left empty, CloudFormation will create a new file system for the stack.
- Select "Y" for DownloadFsxData to automatically populate the FSx for Lustre file system with common sequence databases.
- Select "Y" for CreateG5ComputeEnvironment to create an additional job queue with support for G5 family instances. Note that G5 instances are currently not available in all AWS regions.
If you set the DownloadFsxData parameter to Y, CloudFormation will automatically start a series of Batch jobs to populate the FSx for Lustre instance with a number of common sequence databases. If you set this parameter to N you will instead need to manually populate the file system. Once the CloudFormation stack is in a CREATE_COMPLETE status, you can begin populating the FSx for Lustre file system with the necessary sequence databases. To do this automatically, open a terminal in your notebooks environment and run the following commands from the batch-protein-folding directory:
pip install .
python prep_databases.py
It will take around 5 hours to populate the file system, depending on your location. You can track its progress by navigating to the file system in the FSx for Lustre console.
Please visit [https://github.com/EddyRivasLab/hmmer] for more information about the JackHMMER algorithm.
Please visit [https://github.com/deepmind/alphafold] for more information about the AlphaFold2 algorithm.
The original AlphaFold 2 citation is
@Article{AlphaFold2021,
author = {Jumper, John and Evans, Richard and Pritzel, Alexander and Green, Tim and Figurnov, Michael and Ronneberger, Olaf and Tunyasuvunakool, Kathryn and Bates, Russ and {\v{Z}}{\'\i}dek, Augustin and Potapenko, Anna and Bridgland, Alex and Meyer, Clemens and Kohl, Simon A A and Ballard, Andrew J and Cowie, Andrew and Romera-Paredes, Bernardino and Nikolov, Stanislav and Jain, Rishub and Adler, Jonas and Back, Trevor and Petersen, Stig and Reiman, David and Clancy, Ellen and Zielinski, Michal and Steinegger, Martin and Pacholska, Michalina and Berghammer, Tamas and Bodenstein, Sebastian and Silver, David and Vinyals, Oriol and Senior, Andrew W and Kavukcuoglu, Koray and Kohli, Pushmeet and Hassabis, Demis},
journal = {Nature},
title = {Highly accurate protein structure prediction with {AlphaFold}},
year = {2021},
volume = {596},
number = {7873},
pages = {583--589},
doi = {10.1038/s41586-021-03819-2}
}
The AlphaFold-Multimer citation is
@article {AlphaFold-Multimer2021,
author = {Evans, Richard and O{\textquoteright}Neill, Michael and Pritzel, Alexander and Antropova, Natasha and Senior, Andrew and Green, Tim and {\v{Z}}{\'\i}dek, Augustin and Bates, Russ and Blackwell, Sam and Yim, Jason and Ronneberger, Olaf and Bodenstein, Sebastian and Zielinski, Michal and Bridgland, Alex and Potapenko, Anna and Cowie, Andrew and Tunyasuvunakool, Kathryn and Jain, Rishub and Clancy, Ellen and Kohli, Pushmeet and Jumper, John and Hassabis, Demis},
journal = {bioRxiv}
title = {Protein complex prediction with AlphaFold-Multimer},
year = {2021},
elocation-id = {2021.10.04.463034},
doi = {10.1101/2021.10.04.463034},
URL = {https://www.biorxiv.org/content/early/2021/10/04/2021.10.04.463034},
eprint = {https://www.biorxiv.org/content/early/2021/10/04/2021.10.04.463034.full.pdf},
}
Please visit [https://github.com/aqlaboratory/openfold] for more information about the OpenFold algorithm.
The OpenFold citation is
@software{Ahdritz_OpenFold_2021,
author = {Ahdritz, Gustaf and Bouatta, Nazim and Kadyan, Sachin and Xia, Qinghui and Gerecke, William and AlQuraishi, Mohammed},
doi = {10.5281/zenodo.5709539},
month = {11},
title = {{OpenFold}},
url = {https://github.com/aqlaboratory/openfold},
year = {2021}
}
Please visit [https://github.com/HeliXonProtein/OmegaFold] for more information about the OmegaFold algorithm.
The OmegaFold citation is
@article{OmegaFold,
author = {Wu, Ruidong and Ding, Fan and Wang, Rui and Shen, Rui and Zhang, Xiwen and Luo, Shitong and Su, Chenpeng and Wu, Zuofan and Xie, Qi and Berger, Bonnie and Ma, Jianzhu and Peng, Jian},
title = {High-resolution de novo structure prediction from primary sequence},
elocation-id = {2022.07.21.500999},
year = {2022},
doi = {10.1101/2022.07.21.500999},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/early/2022/07/22/2022.07.21.500999},
eprint = {https://www.biorxiv.org/content/early/2022/07/22/2022.07.21.500999.full.pdf},
journal = {bioRxiv}
}

This architecture uses a nested CloudFormation template to create various resources in a particular sequence:
- (Optional) If existing resources are not provided as template parameters, create a VPC, subnets, NAT gateway, elastic IP, routes, and S3 endpoint.
- (Optional) If existing resources are not provided as template parameters, create a FSx for Lustre file system.
- Download several container images from a public ECR repository and push them to a new, private repository in your account. Also download a .zip file with the example notebooks and other code into a CodeCommit repository.
- Create the launch template, compute environments, job queues, and job definitions needed to submit jobs to AWS Batch.
- (Optional) If requested via a template parameter, create and run a Amazon Lambda-backed custom resource to download several open source proteomic data sets to the FSx Lustre instance.
See CONTRIBUTING for more information.
This project is licensed under the Apache-2.0 License.