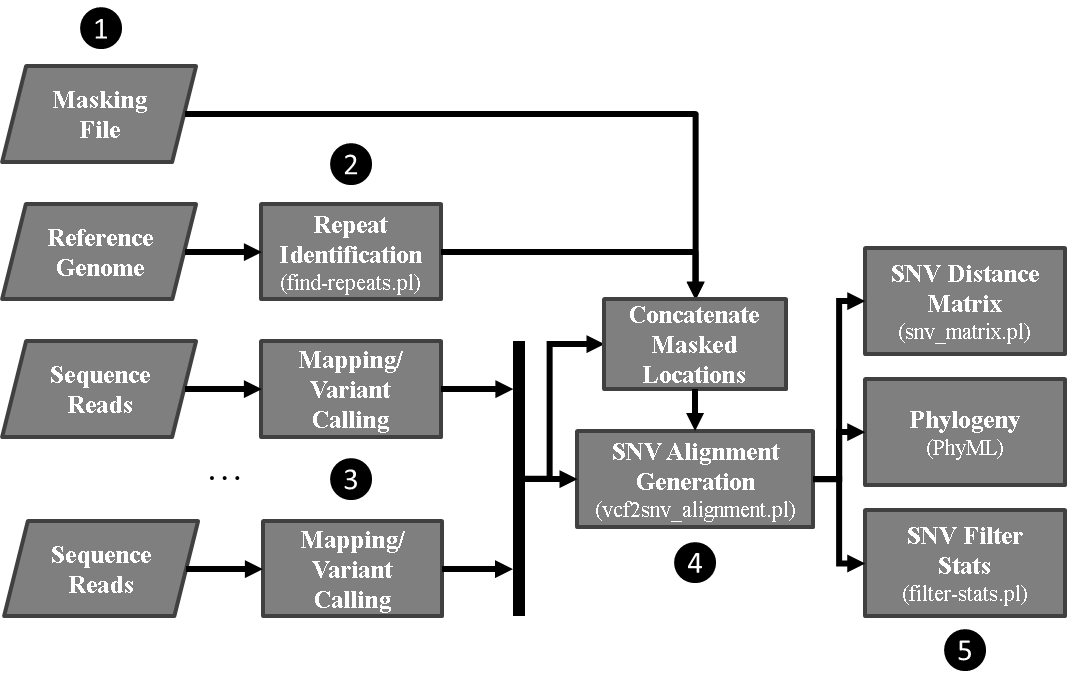
This is a WDL translation of SNVPhyl_Nextflow based on SNVPhyl pipeline. The workflow is optimized for Terra with a few changes:
-
You can enter a full taxon name (e.g., "Escherichia coli") or an NCBI accession number (e.g., "GCF_000005845.2") instead of giving a fasta file for the reference genome. The workflow will download the reference genome automatically. The pipeline hierarchically selects one of them:
fasta file > accession number > taxon name. Although the taxon name is required, it will be ignored when you give an accession number or a reference fasta file. -
Some key parameters (min_coverage, min_mean_mapping, snv_abundance_ratio) are exposed as variables like in the original SNVPhyl pipeline.
-
This pipeline creates an HTML summary report besides the standard outputs of SNVPhyl. The report contains a heatmap-styled SNV matrix and a phylogenetic tree.
Example report
The original SNVPhyl pipeline was written by Aaron Petkau. You can find more information in SNVPhyl paper and documentation. Please keep in mind that this is an indirect adaptation of SNVPhyl from Jill Hagey's SNVPhyl_Nextflow pipeline.
You can install SNVPhyl_Terra from Dockstore as usual.
SNVPhyl_Terra requires a sample set to run!
Parameters:
| Variable | Attribute | Description | Required? |
|---|---|---|---|
| read1 | this.{sampleset name}s.{read1} | required | |
| read2 | this.{sampleset name}s.{read2} | required | |
| samplename | this.{sampleset name}s.{id} | required | |
| taxon | "{string}" | reference taxon | required |
| accession | "{string}" | reference accession number | optional |
| reference | {file} | reference.fasta | optional |
| window_size | {integer} (default: 11) | window size for identifying high-density SNV regions | optional |
| density_threshold | {integer} (default: 2 ) | SNV threshold for identifying high-density SNV regions | optional |
| min_coverage | {integer} (default: 10) | minimum coverage for any given position to be included in the analysis | optional |
| min_mean_mapping | {integer} (default: 30) | minimum mean mapping quality score for all reads in a pileup | optional |
| snv_abundance_ratio | {float} (default: 0.75) | proportion of reads required to support a variant to be included in the analysis | optional |
| colorscale | {string} (default: YlGnBu_r) | background gradient color for SNV matrix. See colormap | optional |
| tree_width | {integer} (default: 600) | phylogenetic tree width | optional |
If you want to use the workflow on your local computer, you can use wf_snvphyl_local.wdl, which is prepared for that purpose. You will need a workflow manager (miniwdl or cromwell) and a container runtime (docker, singularity, etc.) in your path.
Install workflow from GitHub:
git clone https://github.com/Kincekara/SNVPhyl_Terra.git
Prepare inputs:
Create a tab-separated text file (e.g., samples.tsv) including your sample ids and file paths like the below:
sample1 /path_to_sample1_read1.fastq.gz /path_to_sample1_read2.fastq.gz
sample2 /path_to_sample2_read1.fastq.gz /path_to_sample2_read2.fastq.gz
...
Create a JSON file for inputs (e.g., inputs.json)
{
"snvphyl.reference": "/path/to/reference.fasta",
"snvphyl.inputSamplesFile": "/path/to/samples.tsv"
}
Run the workflow by using wf_snvphyl_local.wdl
miniwdl run ~/SNVPhlyl_Terra/workflows/wf_snvphyl_local.wdl -i inputs.json
Petkau, A., Mabon, P., Sieffert, C., Knox, N. C., Cabral, J., Iskander, M., Iskander, M., Weedmark, K., Zaheer, R., Katz, L. S., Nadon, C., Reimer, A., Taboada, E., Beiko, R. G., Hsiao, W., Brinkman, F., Graham, M., & Gary Van Domselaar. (2017). SNVPhyl: a single nucleotide variant phylogenomics pipeline for microbial genomic epidemiology. Microbial Genomics, 3(6). https://doi.org/10.1099/mgen.0.000116
DHQP/SNVPhyl_Nextflow: Nextflow version of SNVPhyl. (2022, April 21). GitHub. https://github.com/DHQP/SNVPhyl_Nextflow