RDBKE is a breakpoint enhancment pipeline for read-depth (RD) based SV callers using deep segmenation model UNet. UNet is used to learn specific patterns of base-wise RDs surrounding known breaking points. It is designed for in-sample and cross-sample applications. More details can be found in the manuscript https://doi.org/10.1101/503649
Old branch DeepIntraSV only contains model-level training and testing. RDBKE branch added the enhancement module for RD-based SV callers (e.g., CNVnator).
-
RDBKE overall pipeline
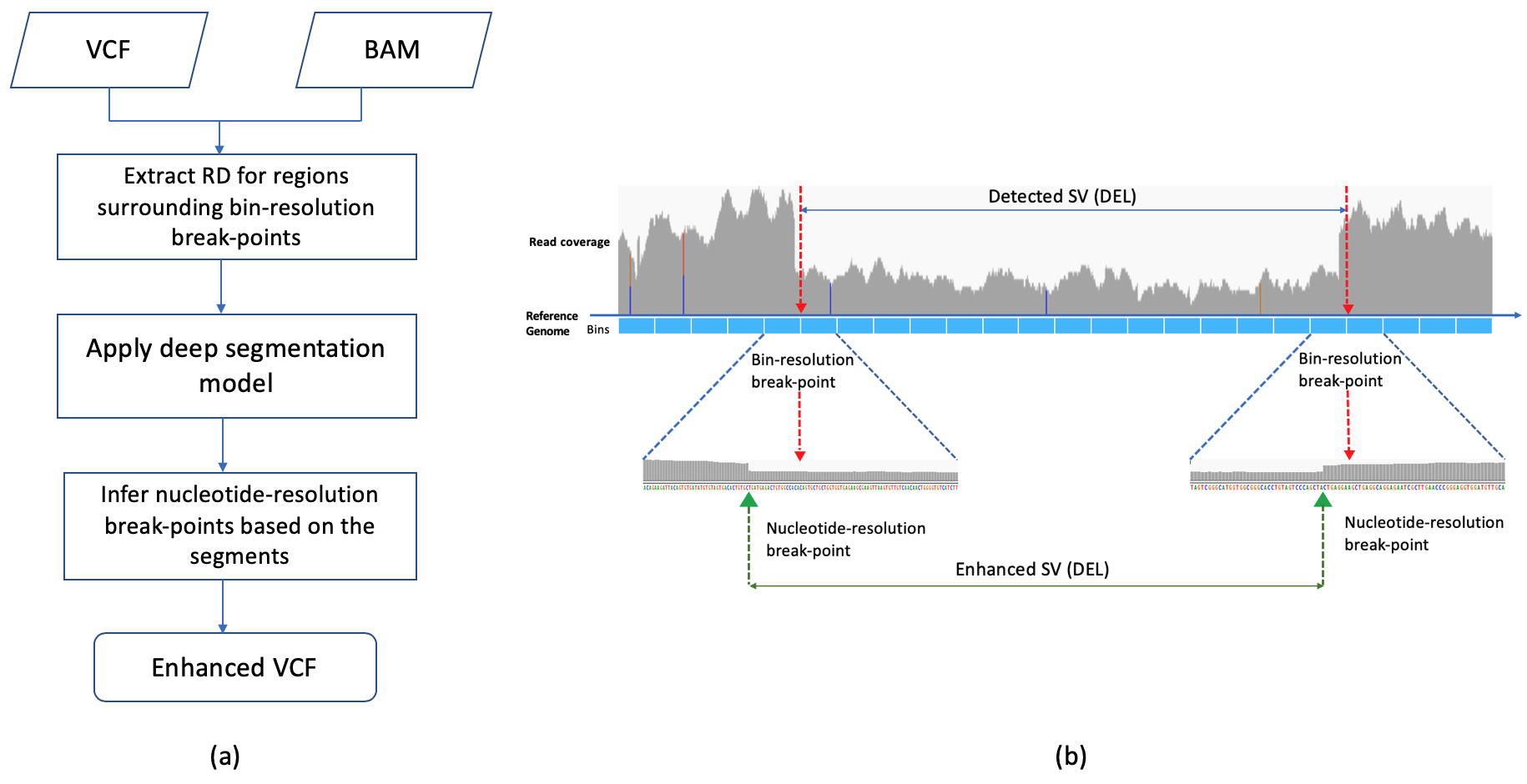
-
Model structure of UNet used for RDBKE:
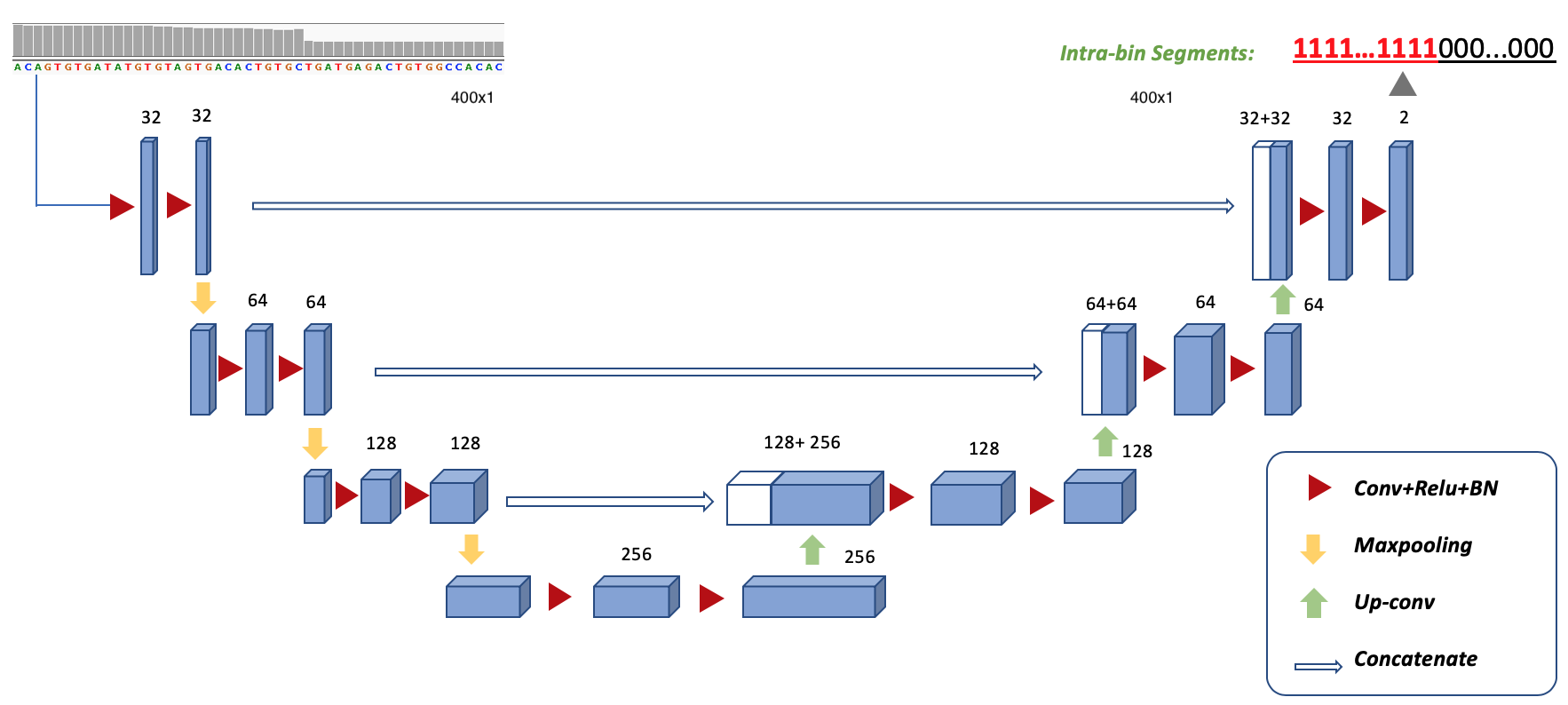


We provide a docker image for running this code
docker pull yaozhong/deep_intra_sv:0.9
- ubuntu 16.04.4
- Tensorflow 1.8.0
- Keras 2.2.4
WORKING_FOLD=<Absolute-PATH>
nvidia-docker run -it --rm -v $WORKING_FOLD:/workspace yaozhong/deep_intra_sv:0.9 bash
The parameter setteing is pre-defined in the code/config.py file.
Some frequently used parameters can be also specifized through command line option (See '-h' option).
- reference files are located in ./data/reference/, which includes required reference files:
- reference genome FASTA file
hs37d5.faand itsindexhs37d5.fa.fai(Please download from [1000genomes](http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/) , and place it in./data/reference/hg19/``) - mappability of 100-mer file
- Encode hg19 blacklist regions
- hg19 Chromesome length
- SV annotation files./data/SV_annotation/ (a python script is provided to parse VCF for SV regions)
# example
sample_name="sample1"
binSize=400
bam_file=<path-of-bam-file>
# SV prediction of a RD-based SV caller, e.g., CNVnator
pVCF=<CNVnator-bin-resolution-prediction-file>
# trained model parameters
model=<path-of-the-trained-model>
# WGS data information, if not cached, it will re-generate.
genomeStat=<path-of-cached-BG-RD-statistics>
output_fold=<restult-saving-path>
# If gold SVs are known, it can be used for evaluating enhancement effect.
gVCF=<path-of-gold-standard-SV>
python eval/enhance_bk.py -d $sample_name -bam $bam_file -b $binSize -gs $genomeStat -mp $model -v $pVCF -o $output_fold
Input are WGS bam file(s) and VCF file(s). Cached train-test data will be first searched according to current parameters, If cache files are not found, the code will process the bam file and cache the data. A multi-core version of pysam is used. By default, all cores will be used to generate RD bins from bam file. For each training data, background statistics of RDs are first calcuated through sampling WGS data. Background statistics of each WGS data will be cached in the same fold of trained model parameters
We provided a default hyperparameters of UNet and CNN in ./experiment/model_param/
Users can make changes of the parameter file or specifiy through command line option.
If no model parameter file is provided, the code will use hyperOpt to search preDefined hyper-parameter spaces based on the train set.
There are two evluation metrics, which are determined through CMD parameter -em (evluation mode)
- in-sample:
-em single - cross-sample:
-em cross
For the in-sample case, only one bam file is required and the data is split into train-test with the following data split -ds option:
- RandRgs: Random split
- CV: cross valdiation
The test-split-proportion can be specified with option -tsp
For the cross-sample case, the second bam file used as the test set is assigned through -d2 option.
# cross-sample
bin_size=400
sample1_name="sample1"
sample2_name="sample2"
BAM=<bam-file-path>
BAM2=<bam2-file-path>
# SVs can be provided in BED or VCF file
VCF=<vcf-file-path>
VCF2=<vcf2-file-path>
model_para="../experiment/model_param/unet_default"
tmp_worksapce="../experiment/tmp_workspace/"
model_save_fold="../experiment/cross_train_model/"
python train.py -b $bin_size -em cross -d $sample1_name -d2 $sample2_name \
-bam $BAM -bam2 $BAM2 -vcf $VCF -vcf2 $VCF2 \
-m UNet -mp $model_para -l dice_loss -tsp 0 -tmp $tmp_worksapce -msf $model_save_fold
# In-sample
bin_size=400
sample1_name="sample1"
BAM=<bam-file-path>
# SVs can be provided in BED or VCF file
VCF=<vcf-file-path>
model_para="../experiment/model_param/unet_default"
tmp_worksapce="../experiment/tmp_workspace/"
model_save_fold="../experiment/single_sample_train_model/"
python train.py -b $bin_size -em single -d $sample1_name \
-bam $BAM -vcf $VCF \
-m UNet -mp $model_para -l dice_loss -tsp 0.8 -tmp $tmp_worksapce -msf $model_save_fold
-
Simulated data:(https://github.com/stat-lab/EvalSVcallers/blob/master/Ref_SV/Sim-A.SV.vcf)
-
NA12878, NA19238, NA19239:(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/integrated_sv_map/ALL.wgs.mergedSV.v8.20130502.svs.genotypes.vcf.gz)
-
HG002:(ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/analysis/NIST_SVs_Integration_v0.6/HG002_SVs_Tier1_v0.6.vcf.gz)
-
COLO829T:(https://zenodo.org/record/3988185/files/truthset_somaticSVs_COLO829.vcf?download=1)
-
NA12878:(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data/NA12878/high_coverage_alignment/NA12878.mapped.ILLUMINA.bwa.CEU.high_coverage_pcr_free.20130906.bam)
-
NA19238:(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data/NA12878/high_coverage_alignment/NA19238.mapped.ILLUMINA.bwa.YRI.high_coverage_pcr_free.20130924.bam)
-
NA19239:(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data/NA12878/high_coverage_alignment/NA19239.mapped.ILLUMINA.bwa.YRI.high_coverage_pcr_free.20130924.bam)
-
HG002:(ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/HG002_NA24385_son/NIST_HiSeq_HG002_Homogeneity-10953946/NHGRI_Illumina300X_AJtrio_novoalign_bams/HG002.hs37d5.60X.1.bam)
-
COLO829T:(ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR275/ERR2752450/COLO829T_dedup.realigned.bam)