![]()
- This work is a derivative of "Transcription factors" by kelvin13, used under CC BY 3.0
Full documentation available at: ReadTheDocs
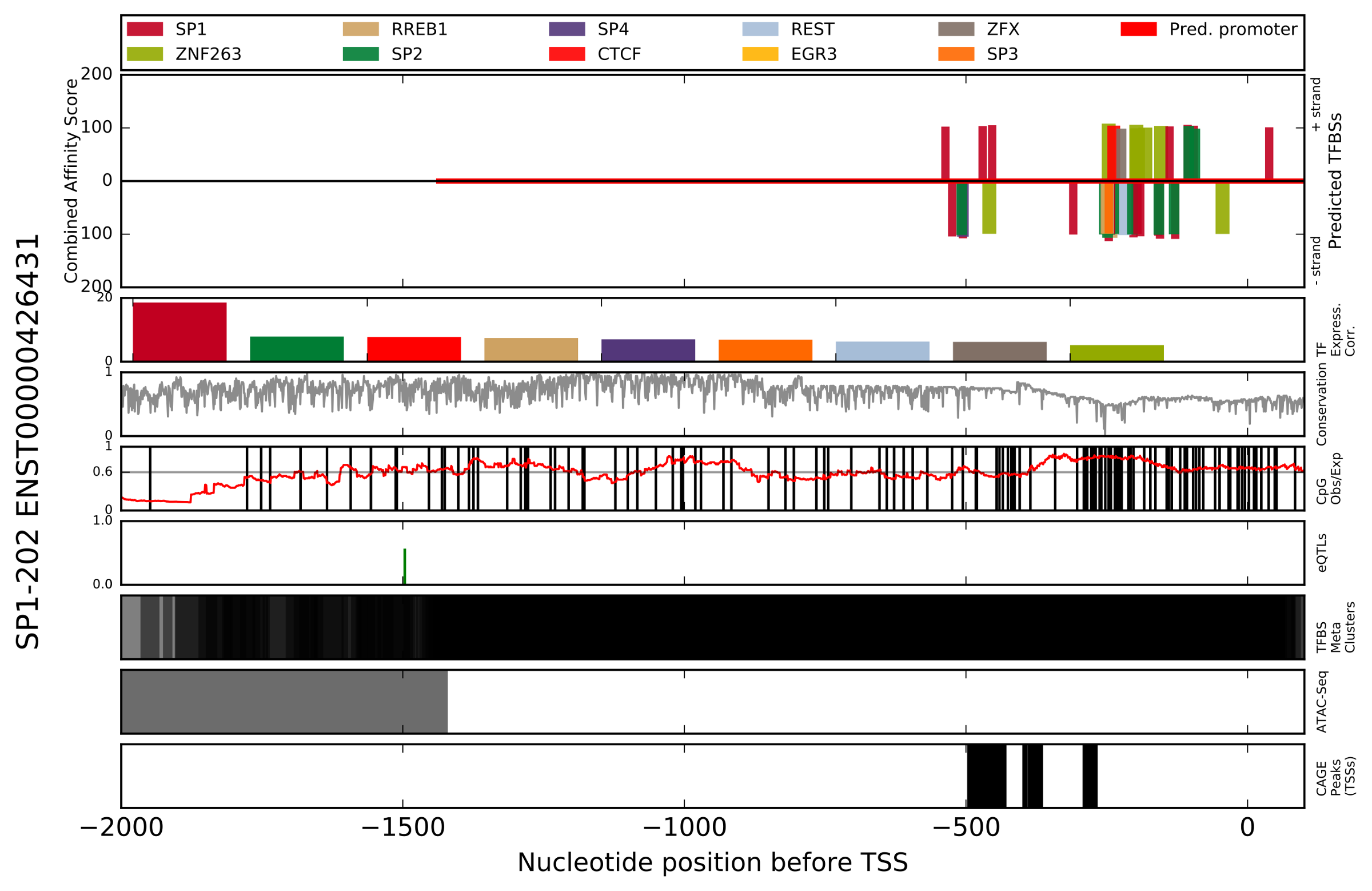
The TFBS footprinting method computationally predicts transcription factor binding sites (TFBSs) in a target species (e.g. homo sapiens) using 575 position weight matrices (PWMs) based on binding data from the JASPAR database. Additional experimental data from a variety of sources is used to support or detract from these predictions:
- DNA sequence conservation in homologous mammal species sequences
- proximity to CAGE-supported transcription start sites (TSSs)
- correlation of expression between target gene and predicted transcription factor (TF) across 1800+ samples
- proximity to ChIP-Seq determined TFBSs (GTRD project)
- proximity to qualitative trait loci (eQTLs) affecting expression of the target gene (GTEX project)
- proximity to CpGs
- proximity to ATAC-Seq peaks (ENCODE project)
- Figure showing top_x_tfs highest scoring (combined affinity score) TFBSs mapped onto target_species promoter (ENSxxxxxxxxxxxx_[species_group].Promoterhisto.svg).
- Original alignment as retrieved from Ensembl (alignment_uncleaned.fasta).
- Cleaned alignment (alignment_cleaned.fasta).
- Regulatory information for the target transcripts user-defined promoter region (regulatory_decoded.json).
- Transcript properties for target transcript (transcript_dict.json).
- All predicted TFBSs for the target species which satisfy p-value threshold (TFBSs_found.all.json).
- All predicted TFBSs for target species which are supported by at least conservation_min predictions in other species, sorted by combined affinity score (TFBSs_found.sortedclusters.csv).
-
Docker
$ docker pull thirtysix/tfbs_footprinting -
Pypi
$ pip install tfbs_footprinting
Predict TFBSs in the promoters any of 1-80,000 human protein coding transcripts in the Ensembl database. TFBS predictions can also be made for 87 unique non-human species (including model organisms such as mouse and zebrafish), present in the following groups:
- 70 Eutherian mammals
- 24 Primates
- 11 Fish
- 7 Sauropsids
View the available Ensembl species groups to plan your analysis: https://rest.ensembl.org/info/compara/species_sets/EPO_LOW_COVERAGE?content-type=application/json
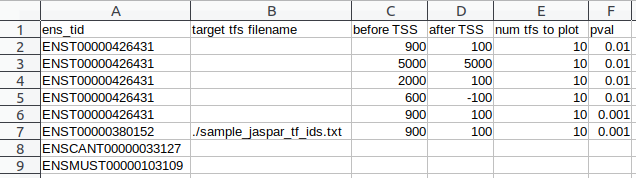
- Option 1: CSV of arguments
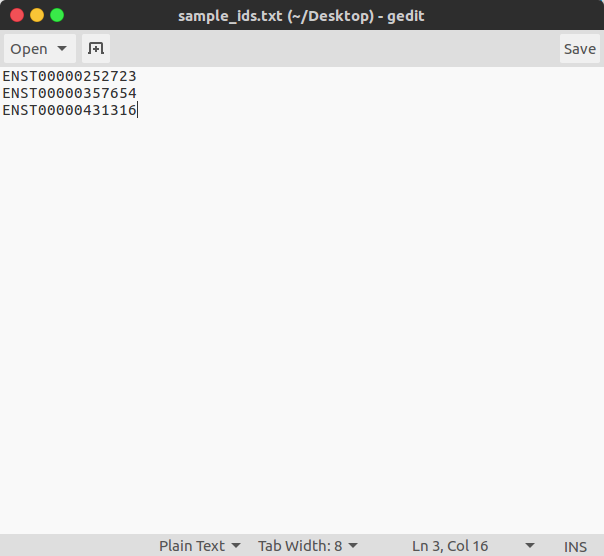
- Option 2: Simple text-file of Ensembl Transcript IDs
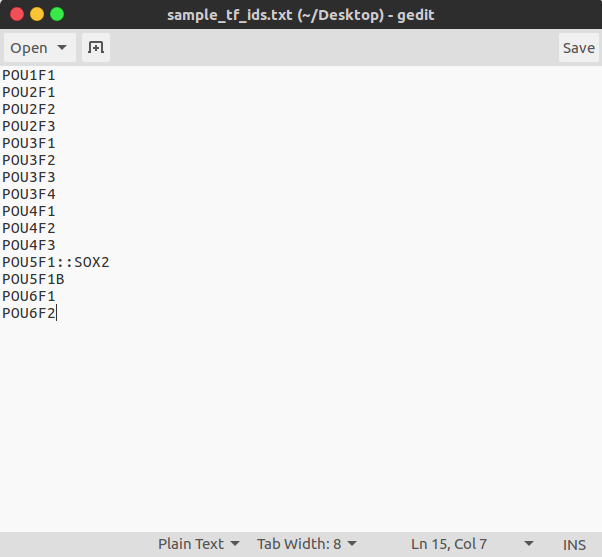
- File of Jaspar TF IDs (Not required)
-
Run the sample analysis using a .csv of arguments:
$ tfbs_footprinter -t PATH_TO/sample_analysis/sample_analysis_list.csv -
Run the sample analysis using a .txt of Ensembl transcript ids, and minimal arguments:
$ tfbs_footprinter -t PATH_TO/sample_analysis/sample_ensembl_ids.txt
-
Run the sample analysis using a .txt of Ensembl transcript ids, and all arguments:
$ tfbs_footprinter -t PATH_TO/sample_analysis/sample_ensembl_ids.txt -tfs PATH_TO/sample_analysis/sample_jaspar_tf_ids.txt -s homo_sapiens -g mammals -e low -pb 900 -pa 100 -tx 10 -update
$ tfbs_footprinter -update
-
Within Docker we first need to mount a volume so that the results of the analyis can be viewed on our host computer. It is recommended that you create an empty directory on your host computer:
$ docker run -v /ABSOLUTE_PATH_TO/EMPTY_DIR_ON_HOST:/home/sample_analysis/tfbs_results -it thirtysix/tfbs_footprinting bash -
Then we move into the pre-existing sample analysis directory in the Docker container to perform the analysis there so that the results generated there will automatically appear in the designated location on our host computer:
$ cd ./sample_analysis -
Then we can run the sample analysis in Docker in the same way that we would normally use tfbs_footprinter (above), e.g. using a .csv of arguments:
$ tfbs_footprinter -t ./sample_analysis_list.csv
-
Or (again, as above) using a .txt of Ensembl transcript ids, and minimal arguments:
$tfbs_footprinter -t ./sample_ensembl_ids.txt -
Or (again, as above) using a .txt of Ensembl transcript ids, and multiple arguments:
$ tfbs_footprinter -t ./sample_ensembl_ids.txt -tfs ./sample_jaspar_tf_ids.txt -s homo_sapiens -g mammals -e low -pb 900 -pa 100 -tx 10 -o ./tfbs_results -update
-
Within Docker we first need to mount a volume so that we can load your analysis files from your host computer into docker AND save the results of the analysis on our host computer:
$ docker run -v /ABSOLUTE_PATH_TO/DIR_ON_HOST/CONTAINING_ANALYSIS_FILES:/home/analysis_dir -it thirtysix/tfbs_footprinting bash -
Then we move into your analysis directory in the Docker container to perform the analysis there so that the results generated there will automatically appear in the designated location on our host computer:
$ cd ./analysis_dir -
Then we can run the sample analysis in Docker in the same way that we would normally use tfbs_footprinter (above), e.g. using a .csv of arguments:
$ tfbs_footprinter -t ./USER_TABLE_OF_ENSEMBL_IDS_AND_ARGS.csv -
Or (again, as above) using a .txt of Ensembl transcript ids, and minimal arguments:
$ tfbs_footprinter -t ./USER_LIST_OF_ENSEMBL_IDS.txt
-
Or (again, as above) using a .txt of Ensembl transcript ids, and multiple arguments:
$ tfbs_footprinter -t ./USER_LIST_OF_ENSEMBL_IDS.txt -tfs ./USER_LIST_OF_TF_NAMES.txt -s homo_sapiens -g mammals -e low -pb 900 -pa 100 -tx 10 -o PATH_TO/Results/ -update
- --help, -h show this help message and exit
- --t_ids_file, -t
Required for running an analysis. Location of a file containing Ensembl target_species transcript ids. Input options are either a text file of Ensembl transcript ids or a .csv file with individual values set for each parameter. - --tf_ids_file, -tfs
Optional: Location of a file containing a limited list of Jaspar TFs to use in scoring alignment (see sample file tf_ids.txt at https://github.com/thirtysix/TFBS_footprinting) [default: all Jaspar TFs] - --target_species, -s [default: "homo_sapiens"] - Target species (string), options are located at https://github.com/thirtysix/TFBS_footprinting/blob/master/README.md#6-species. Conservation of TFs across other species will be based on identifying them in this species first.
- --species_group, -g ("mammals", "primates", "sauropsids", or "fish") [default: "mammals"] - Group of species (string) to identify conservation of TFs within. Your target species should be a member of this species group (e.g. "homo_sapiens" and "mammals" or "primates"). The "primates" group does not have a low-coverage version. Groups and members are listed at https://github.com/thirtysix/TFBS_footprinting/blob/master/README.md#6-species.
- --coverage, -e
("low" or "high") [default: "low"] - Which Ensembl EPO alignment of species to use. The low coverage contains significantly more species and is recommended. The primate group does not have a low-coverage version. - --promoter_before_tss, -pb (0-100,000) [default: 900] - Number (integer) of nucleotides upstream of TSS to include in analysis. If this number is negative the start point will be downstream of the TSS, the end point will then need to be further downstream.
- --promoter_after_tss, -pa (0-100,000) [default: 100] - Number (integer) of nucleotides downstream of TSS to include in analysis. If this number is negative the end point will be upstream of the TSS. The start point will then need to be further upstream.
- --top_x_tfs, -tx (1-20) [default: 10] - Number (integer) of unique TFs to include in output .svg figure.
- --pval, -p P-value (float) for determine score cutoff (range: 0.1 to 0.0000001) [default: 0.01]
- --exp_data_update, -update Download the latest experimental data files for use in analysis. Will run automatically if the "data" directory does not already exist (e.g. first usage).
Iterate through each user provided Ensembl transcript id:
- Retrieve EPO aligned orthologous sequences from Ensembl database for user-defined species group (mammals, primates, fish, sauropsids) for promoter of user-provided transcript id, between user-defined TSS-relative start/stop sites.
- Edit retrieved alignment:
- Replace characters not corresponding to nucleotides (ACGT), with gaps characters "-".
- Remove gap-only columns from alignment.
- Generate position weight matrices (PWMs) from Jaspar position frequency matrices (PFMs).
- Score target species sequence using either all or a user-defined list of PWMs.
- Keep predictions with a log-likelihood score greater than score threshold corresponding to p-value of 0.001, or user-defined p-value.
- When experimental data is available for the target species, score each of the following for the target sequence region:
- DNA sequence conservation in homologous mammal species sequences
- proximity to CAGE-supported transcription start sites (TSSs)
- correlation of expression between target gene and predicted transcription factor (TF) across 1800+ samples
- proximity to ChIP-Seq determined TFBSs (GTRD project)
- proximity to qualitative trait loci (eQTLs) affecting expression of the target gene (GTEX project)
- proximity to CpGs
- proximity to ATAC-Seq peaks (ENCODE project)
- Compute 'combined affinity score' as a sum of scores for all experimental data.
- Sort target_species predictions by combined affinity score, generate a vector graphics figure showing the top 10 (or user-defined) unique TFs mapped onto the promoter of the target transcript, and additional output as described below.
The promoter region of any Ensembl transcript of any species within any column can be compared against the other members of the same column in order to identify a conserved binding site of the 575 transcription factors described in the Jaspar database. The Enredo-Pecan-Ortheus pipeline was used to create whole genome alignments between the species in each column. 'EPO_LOW' indicates this column also contains genomes for which the sequencing of the current version is still considered low-coverage. Due to the significantly greater number of species, we recommend using the low coverage versions except for primate comparisons which do not have a low coverage version. This list may not fully resp
| EPO_LOW mammals | EPO_LOW fish | EPO_LOW sauropsids | EPO mammals | EPO primates | EPO fish | EPO sauropsids |
|---|---|---|---|---|---|---|
| ailuropoda_melanoleuca | astyanax_mexicanus | anas_platyrhynchos | bos_taurus | callithrix_jacchus | danio_rerio | anolis_carolinensis |
| bos_taurus | danio_rerio | anolis_carolinensis | callithrix_jacchus | chlorocebus_sabaeus | gasterosteus_aculeatus | gallus_gallus |
| callithrix_jacchus | gadus_morhua | ficedula_albicollis | canis_familiaris | gorilla_gorilla | lepisosteus_oculatus | meleagris_gallopavo |
| canis_familiaris | gasterosteus_aculeatus | gallus_gallus | chlorocebus_sabaeus | homo_sapiens | oryzias_latipes | taeniopygia_guttata |
| cavia_porcellus | lepisosteus_oculatus | meleagris_gallopavo | equus_caballus | macaca_mulatta | tetraodon_nigroviridis | |
| chlorocebus_sabaeus | oreochromis_niloticus | pelodiscus_sinensis | felis_catus | pan_troglodytes | ||
| choloepus_hoffmanni | oryzias_latipes | taeniopygia_guttata | gorilla_gorilla | papio_anubis | ||
| dasypus_novemcinctus | poecilia_formosa | homo_sapiens | pongo_abelii | |||
| dipodomys_ordii | takifugu_rubripes | macaca_mulatta | ||||
| echinops_telfairi | tetraodon_nigroviridis | mus_musculus | ||||
| equus_caballus | xiphophorus_maculatus | oryctolagus_cuniculus | ||||
| erinaceus_europaeus | ovis_aries | |||||
| felis_catus | pan_troglodytes | |||||
| gorilla_gorilla | papio_anubis | |||||
| homo_sapiens | pongo_abelii | |||||
| ictidomys_tridecemlineatus | rattus_norvegicus | |||||
| loxodonta_africana | sus_scrofa | |||||
| macaca_mulatta | ||||||
| microcebus_murinus | ||||||
| mus_musculus | ||||||
| mustela_putorius_furo | ||||||
| myotis_lucifugus | ||||||
| nomascus_leucogenys | ||||||
| ochotona_princeps | ||||||
| oryctolagus_cuniculus | ||||||
| otolemur_garnettii | ||||||
| ovis_aries | ||||||
| pan_troglodytes | ||||||
| papio_anubis | ||||||
| pongo_abelii | ||||||
| procavia_capensis | ||||||
| pteropus_vampyrus | ||||||
| rattus_norvegicus | ||||||
| sorex_araneus | ||||||
| sus_scrofa | ||||||
| tarsius_syrichta | ||||||
| tupaia_belangeri | ||||||
| tursiops_truncatus | ||||||
| vicugna_pacos |