This tutorial will hopefully teach you enough about BED files and Bedtools to begin conducting genomic and sequencing analysis.
Bedtools is a huge package with a wide variety of commands and functions which I don't personally know completely. More important than memorizing every single command and its usage is to instead become adapt at navigating the bedtools documentation and become a pro at using resources like google and peers to address data analysis challenges. I hope that the following tutorial will provide these skills which will continue to be integral to your future bioinformatics projects.
BED files are essentially text files which store genomic positional information. They are tab-delimited files, just like TSVs, except they contain no header row.
Because BED files do not have any official specification, the data per column is largely subjective. However, there are 3 obligatory columns which must always contain the same information.
- Column 1 must always contain the chromosome or scaffold identity (chr1, chrY, scaffold17532....)
- Column 2 must always contain the start coordinate
- Column 3 must always contain the end coordinate. This end value is non-inclusive
Besides these 3 columns, additional columns of other information can be inputted at the discretion of the user. However, in general the column descriptions popularised by the UCSC genome browser are recommended but not required (see the descriptions here).
BED files are often used to list important genomic positions in a smaller format than including positions and sequence data in Fasta, BigWig, BAM/SAM/CRAM, and other such formats. They are incredibly useful for highlighting specific regions of a genome or for inputting into other software packages to get greater information on the described genomic region. For example, a list of putative enhancers can be denoted in a BED file as such:
chr1 18537 18754 enhancer1
chr2 7583726 7594726 enhancer2
NOTE these BED files are completely different from BED files used in the PLINK software package. Computational biology file formats are disgusting :'(.
Bedtools is a software package which allows for the easy manipulation and comparision of BED files. For example, lets say we have two BED files for alternatively spliced exons in two cell populations. How can we determine which exons are in both cell populations? Bedtools makes such operations incredibly fast and simple.
Bedtools can be installed here. Additionally, a python version of Bedtools called pyBedTools can also be installed from here. pyBedTools is useful if you want to run custom python scripts on BED files, but generally not needed.
Here is a list of the commands which I personally use the most:
| Command | Description | example | further reading |
|---|---|---|---|
intersect |
find all regions of overlap between two BED files | bedtools intersect -a 1.bed -b 2.bed |
link |
sort |
sort all intervals in a BED file, by default by chromosome then position | bedtools sort -i 1.bed |
link |
merge |
combine overlapping regions of a BED file into a single region | bedtools merge -i 1.bed |
link |
Sorting is an incredibly useful tool, both a necessary component of certain Bedtool command such as merge, and also generaly works to speed up calculations and decrease RAM usage. So in general, always try and sort your files first, especially if they are large files.
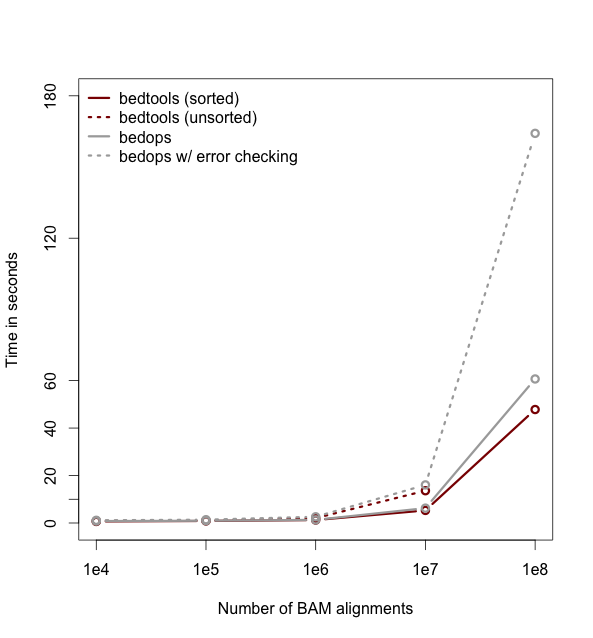
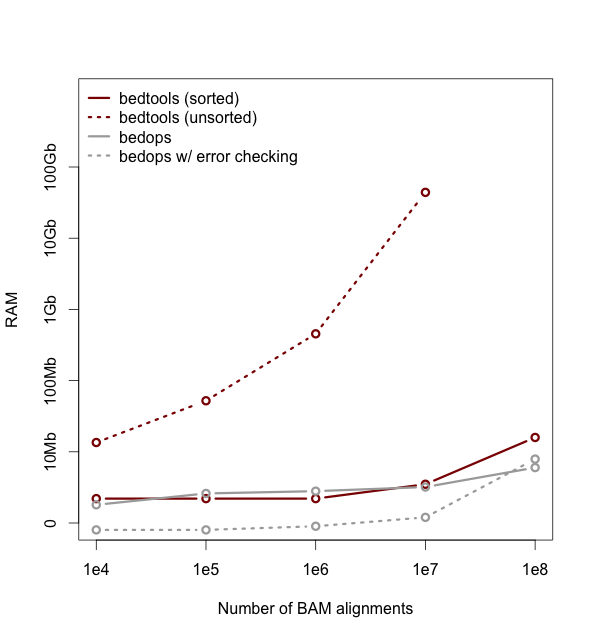
A full list of bedtools commands can be found here and a very in-depth tutorial on using bedtools can also be found here.
Alright, now lets try out some bedtool commands.
- First lets download some BED files. Download two h3k27ac CHIP-seq bed narrowPeak files from different tissue types of the same species and genomic library (hg19, mm9, ...) from the encodeproject.org.
- After downloading, compare the two bed files. What sequences do they share in common? What sequences are unique?
- Lets say we want to find the overlapping regions of these two bed files. However, we only want the overlapping sequence which is longest between the two samples. For example, if one BED file has a region of
chr1 1 10and another has a region ofchr1 5 20, only output the second interval since it is the longest. - We want to find enhancers specific to a single tissue type. How would you go about doing so? Try and create a reasonable list of enhancers specific to a single tissue type from BED files from ENCODE.
- Nice, now we have a list of putative enhancers. One common method of identifying enhancers is via level of conservation. How conserved are your enhancers? Convert your BED sequences to another species genome library using liftover. Then, try finding overlaps between these converted regions and a downloaded set of h3k27ac peaks from ENCODE for your interested tissue type and species you are comparing it to. eg. If you have a list of enhancers in human lung tissue aligned to hg19, convert it to mm9 and compare it to h3k27ac peaks in mouse lung tissue also aligned to mm9.
- Which of these enhancers occur in intronic regions?