non-optimal scenarios benchmarking of sc(n/multiome)RNA-seq hashing demultiplex tools
Some hashing data has just been rushed to the emergency room. It's clearly not doing well -- the data is messy, maybe not even bimodal! So can it be rescued? Which tool(s), often designed and optimized for idealized situations, should be called upon?
- Iterate over different demultiplexing tools, given the same input files
- Summarize accuracy and other metrics, given ground truth and cell type info
- Generate basic QC for hashing data, and suggest appropriate demultiplexing tool
- Present a collection of real data with edge non-optimal cases and ground truth (freemuxlet genotyping, sex-specific gene expression, unique expression profiles, etc) for benchmarking
- Limited sequencing depth - (
test_lowreads,get_saturation) - Tag count distribution not bi-modal - (
test_bimodal) - High background contamination from one tag - (
test_background) - Cell type bias for staining and peak count - (
test_typebias)
Overall wrapper test_all
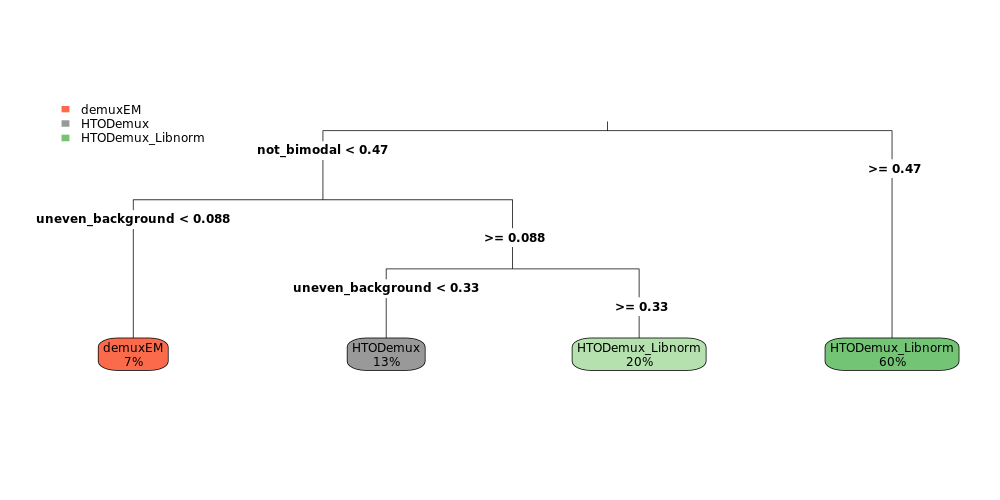
We often observe a cell-type bias for hashtag (feature barcoding) quantification and demultiplex results. In brief, if certain cell types attach to the hashtags more readily than others (more surface targets, larger size, etc), the peak intensities and thresholds for these cell types would naturally be different, which affects the single threshold calling results of HTODemux. Adding a simple library size normalization to the default CLR transformation in our experience generally helps improve demultiplex performance, with minimal impact to runtime or resources. This can be called with run_HTODemux_Libnorm from input files, and HTODemux_Libnorm for Seurat objects as input.
- Seurat default functions
HTODemuxandMULTIseqDemuxare generally effective, but are prone to demultiplex certain cell types less effectively (for instance in the brain, neurons are consistently demuxed at higher singlet rate than oligodendrocytes, etc). demuxEM, now part of Cumulus and Pegasus, tends to do better at avoiding singlet demux differences among cell types. However, it struggles handling uneven background from the hashtags (~5x diff from high/low).HTODemux_Libnormmodification outperformsHTODemuxandMULTIseqDemuxwhen data is not cleanly bi-modal (and actively does worse if it is), while also attenuating potential cell type bias issue.HTODemux_Libnormalso suffers the sharpest dropoff in effectiveness with low tag counts (~20 UMI counts per cell median).- Therefore, no solution fits all situations. Researchers should be careful to explore and visualize hashing data (with help from the QC functions provided here) and choose appropriate remedies.