Expand the shared genespace between plant species to improve integration!
Go to the website »
Build a new coexpression network
·
View the Preprint
·
Lab Website and Contact Info
Table of Contents
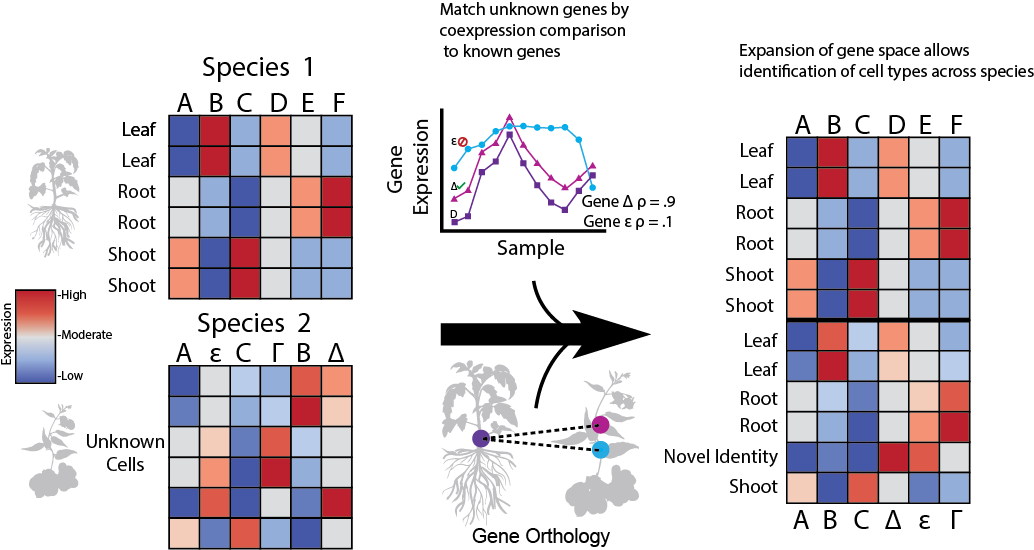
Welcome to EPIPHITES! This project was inspired by a late night trying to help our tireless collaborators. After many months of attempting to get integration of Arabidopsis and Maize data working, we realized that the tiny number of 1-1 genes was majorly impacting integration. To deal with this, we use conservation of coexpression to identify highly similar gene pairs across species, and add them to the 1-1 gene space. Why does this improve integration?
Here's why:
- All gene expression is highly correlated - gene expression does not look like 20k independent values
- This correlated expression underlies all single cell analysis - correlated expression enables the clustering that facilliates single cell
- Coexpression captures both function and regulation, finding good matches for genes across species. Additionally, the one to one genes are not perfect matches either, as they've evovled between species and taken on new functions. This gives flexibility to allow mismatches in both the true 1-1 genes and our coexpression proxies
The best part about EPIPHITES is how easy it is to drop into your existing workflow and see if it improves your results. All you need to do to try it out is pop-over to the Shiny app, download the genelist for your species pair of interest, and give integration a shot with your exisiting workflow!
This will explain how to get started if you are interested in integrating high dimensional data between two of the following species: Zea mays, Oryza sativa, Arabidopsis thaliana, Malus domestica, Sorghum bicolor, Vitis vinifera, Brassica rapa, Glycine max, Medicago truncatula, Nicotiana tabacum, Solanum tuberosum, Brachypodium distachyon, Solanum lycopersicum. The coexpression proxies from these species are identified by comparing coexpression networks generated from thousands of bulk samples. If instead, you'd like to generate new coexpression proxies between two different species, pop on down to the section below.
Using coexpression proxies between species for which we have precalculated them is very easy! First, pop over to our Shiny webpage, right over here! From here, you're going to pick the two species you need to integrate between, and what stringency threshold you'd like the coexpression proxies at. What is the stringency threshold? Its basically how similar we require the genes coexpression profile to be in order to call them as coexpression proxies, as well as how much better of a match the genes need to be than any other potential match. The stringent threshold requires the tightest match, and the lenient threshold requires a much lower match. After downloading your list of genes, its easy to drop it into your integration workflow. First, you'll want to make a copy of the original data, and also move a version of the data to .raw (or to a backup assay if using Seurat). Next, you replace the gene names in one species with the matched gene from the other species based on the downloaded file, and then drop the non-matched genes from each dataset. Next, you'll do any preprocessing required for your chosen integration (for example, concatanating the files and setting a batch variable) and then run your chosen integration technique. Following integration, we recommend Metaneighbor for evaluating your integration quality! Please don't rely only on a 2 dimensional projection (eg. UMAP/TSNE) to evaluate integration.
If you are integrating with a species for which we don't have precalculated data, you can pop over to this Python notebook in order to be walked through the steps. To briefly describe the workflow, we first generate a coexpression network by finely clustering the single cell data, partitioning cells into groups of ~10 highly similar cells. Then, using each of these groups as a sample, we generate a coexpression network. In comparisions to Arabidopsis bulk data, we find that coexpression networks generated this way are of very good quality, even when generated from only root tissue. Now that we have a coexpression network, we are able to move to the standard coexpression pipeline. Reading in a gene orthology, we calculate the conservation of coexpression between all gene members in the same groups. Picking a set of thresholds (we suggest moderate), we then trim down the groups to identify coexpression proxies. This output is then used like any of our pregenerated lists to integrate the two species.
Distributed under the MIT License. See LICENSE.txt for more information.
Michael Passalacqua- @nomadicscience - passala@cshl.edu
Jesse Gillis- Faculty Page - jesse.gillis@utoronto.ca
This project is only possible because of my very supportive lab and collaborators. The Jackson and Lippman Lab at CSHL, the Birnbaum Lab at NYU, and my fantastic mentors and inspirations Hamsini, and Maggie.