Ancestral Cost is a tool for validating multiple sequence alignments prior to performing ancestral sequence reconstruction.
It checks for each position in a given ancestor that the presence of ancestral content implied to be there by a given alignment and tree is not substantially less parsimonious then the alternative of not having ancestral content there.
$ pip install ancestralcost $ git clone https://github.com/gabe-foley-thesis/ancestralcost
$ cd ancestralcost
$ python setup.py install$ ancestralcost -a <alignment> -t <tree>Before performing ancestral sequence reconstruction (ASR) we can recognise that a multiple sequence alignment implies that every aligned column should have a common ancestor.
Ancestral Cost checks that for every ancestral position that is implied by a given alignment and tree the parsimony cost of having ancestral content there isn't far greater than not having ancestral content.
Ancestral Cost is intended to be run before ASR in order to validate alignments and trees. It highlights positions that may be erroneously aligned.
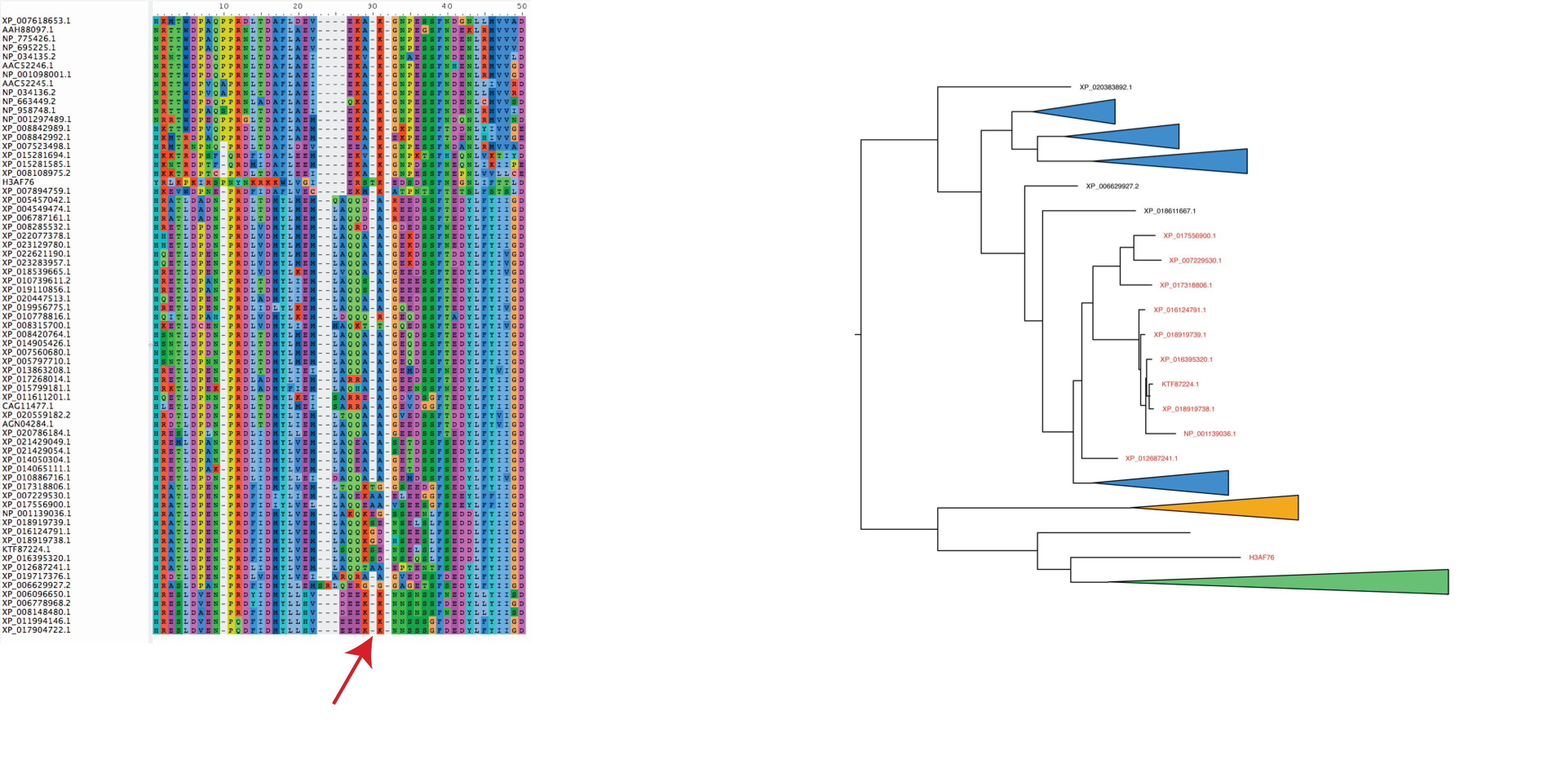
If an alignment suggests two positions should be aligned but they are only present in distant clades then they shouldn't be one column but split into two columns. Failing to do this will influence ancestors that are predicted at these positions.
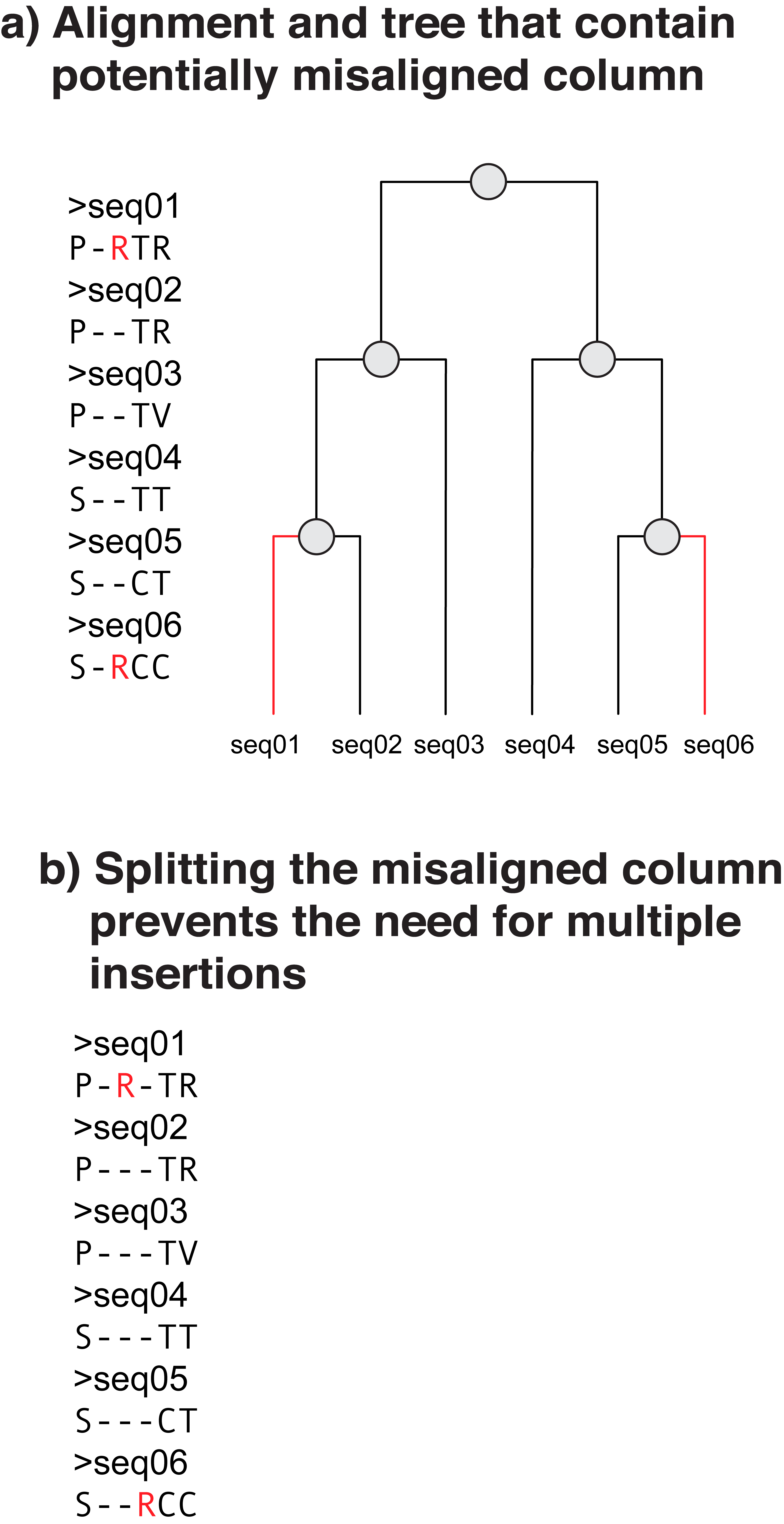
First Ancestral Cost calculates all of the positions required to be there. In the example this is done by simply looking at the highest ancestral position implied by each column. From the example, N3 is the only ancestral node that has content at each of the four alignment positions, all of the other nodes have content at three alignment positions.

It then calculates the parsimony cost for each implied position and reports on the cost of content being present and cost of content being absent.
This allows users to filter on particularly informative sites or particularly large discrepencies in parsimony scores.
The intention is to look at the positions identified by Ancestral Cost and potentially amend the multiple sequence alignment as a result.
-a Path to alignment
-t Path to phylogenetic tree
-n Node to return cost for (default is root)
-p Just return the positions required to be there
-f Return all ancestors as a FASTA file
-to Write out the ancestor tree