





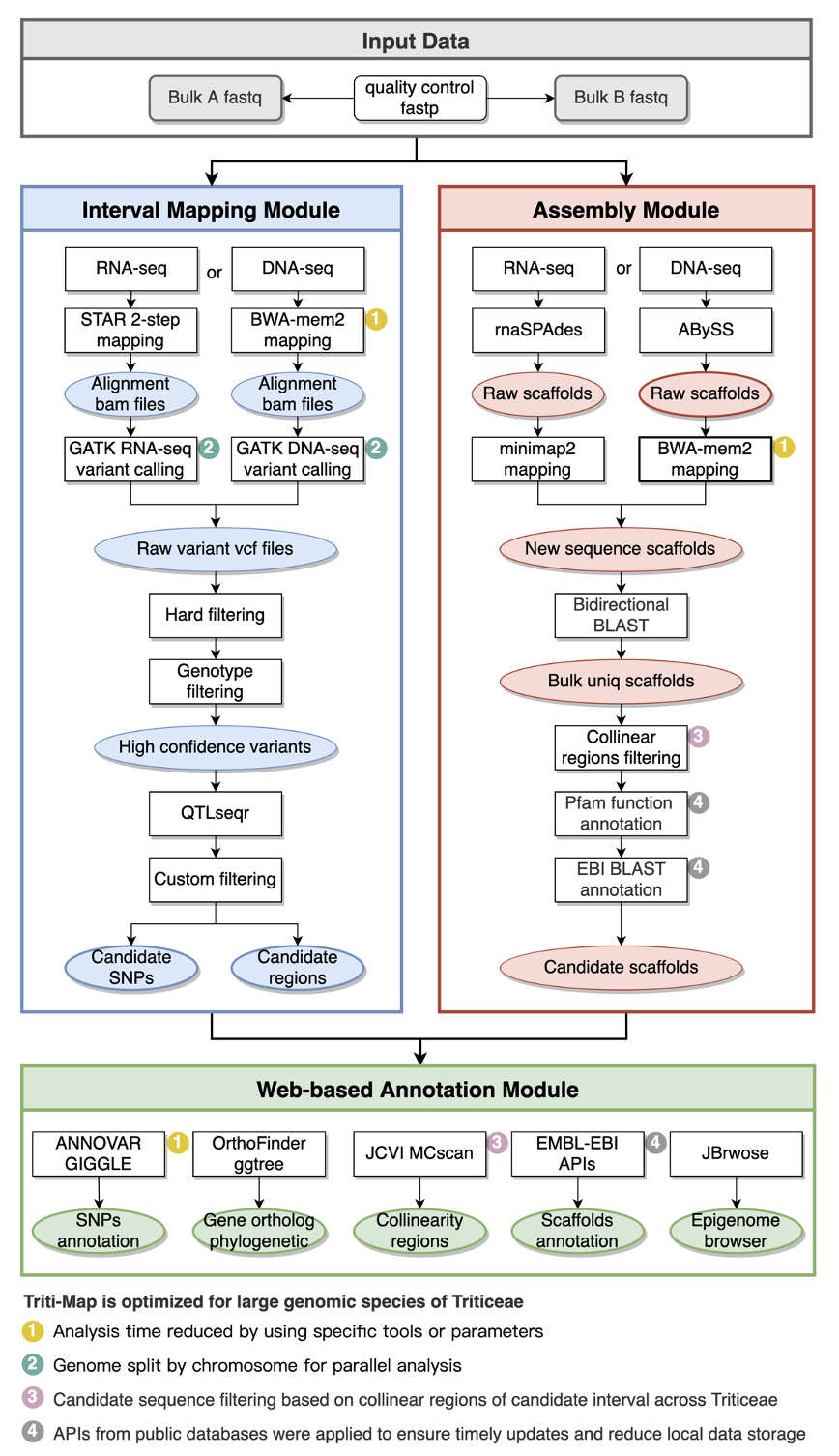
Triti-Map is a Snakemake-based pipeline for gene mapping in Triticeae, which contains a suite of user-friendly computational packages and web-interface integrating multi-omics data from Triticeae species including genomic, epigenomic, evolutionary and homologous information.
Triti-Map could efficiently explore trait-related genes or functional elements not present in the reference genome and reduce the time and labor required for gene mapping in large genome species.
More thorough information and explanations are provided in the Triti-Map Wiki.

Triti-Map ptimization steps to address specific challenges of Triticeae gene-mapping
First, to install Triti-Map you need a UNIX environment contains Bioconda. See how to install Conda
# create new environment and install Triti-Map
conda create -c conda-forge -c bioconda -n tritimap tritimap
# activate Triti-Map environment
conda activate tritimap
# test Triti-Map
tritimap --helpYou can also use Triti-Map via Docker. See how to install Docker, then download and run this image using the following commands:
# docker pull command
docker pull fei0810/tritimap:v0.9.7
# run docker
docker run -i -t fei0810/tritimap:v0.9.7 /bin/bash# download Triti-Map
git clone https://github.com/fei0810/Triti-Map.git
cd Triti-Map
# install Triti-Map
python setup.py installWhen using source code for installation, you need to install other dependencies of Triti-Map by yourself. You can view Triti-Map dependent software via tritimap_env.yaml
Downloading the genome and annotation files you need. Here are some links to download the genome of the Triticeae species.
- Triticum aestivum
- Triticum turgidum
- Triticum dicoccoides
- Hordeum vulgare
- Aegilops tauschii
- Triticum urartu
Building GATK and samtools index file
# for example
# GATK index
gatk CreateSequenceDictionary -R /genome/path/genome.fasta
# samtools index
samtools faidx /genome/path/genome.fastaDNA-seq data use bwa-mem2。
# for example
bwa-mem2 index /genome/path/genome.fastaRNA-seq data use STAR。
# for example
STAR --runThreadN 30 --runMode genomeGenerate \
--genomeDir /star/index/path/genome_star \
--genomeFastaFiles /genome/path/genome.fasta \
--sjdbOverhang 100 \
--sjdbGTFfile /anntotaion/path/genome.gtf \
--genomeChrBinNbits 18 \
--limitGenomeGenerateRAM 50805727274# generate configuration file in current directory
tritimap init
#Or generate configuration file in running directory
tritimap init -d /your/work/pathWhen tritimap init is run successfully, the working directory will generate three configuration files that you need to modify.
config.yaml: Triti-Map configuration filesample.csv: Sample information fileregion.csv: Chromosome region file used to filter the raw results (required only when runningAssembly Modulealone)
The Triti-Map wiki contains detailed information about the meaning and usage of each parameter in the configuration file.
conda activate tritimap
# running directory
cd /your/work/path
# three types of analysis method
# running both Interval Mapping Module and Assembly Module
tritimap run -j 30 all
# only running Interval Mapping Module
tritimap run -j 30 only_mapping
# only running Assembly Module
tritimap run -j 30 only_assembly
Triti-Map supports three types of analysis method.
tritimap run -j 30 only_mapping: If you only need to identify trait association intervals and mutations, then run the Interval Mapping Module.tritimap run -j 30 only_assembly: If you only need to identify trait association new genes, then run the Assembly Module.tritimap run -j 30 all: run the Interval Mapping Module and the Assembly Module together.
Note: Triti-Map pipeline may take a long time to run(1 to 2 days). The screen command is useful for the cases when you need to start a long-running process. Learn more about GNU Screen.
A complete catalog of Triti-Map results is shown below.
├── results
│ ├── 01_cleandata
│ ├── 02_mergedata
│ ├── 03_mappingout
│ ├── 04_GATKout
│ ├── 05_vcfout
│ ├── 06_regionout
│ ├── 07_assembleout
│ ├── logs
You can learn about the results generated by Triti-Map in the Triti-Map wiki.
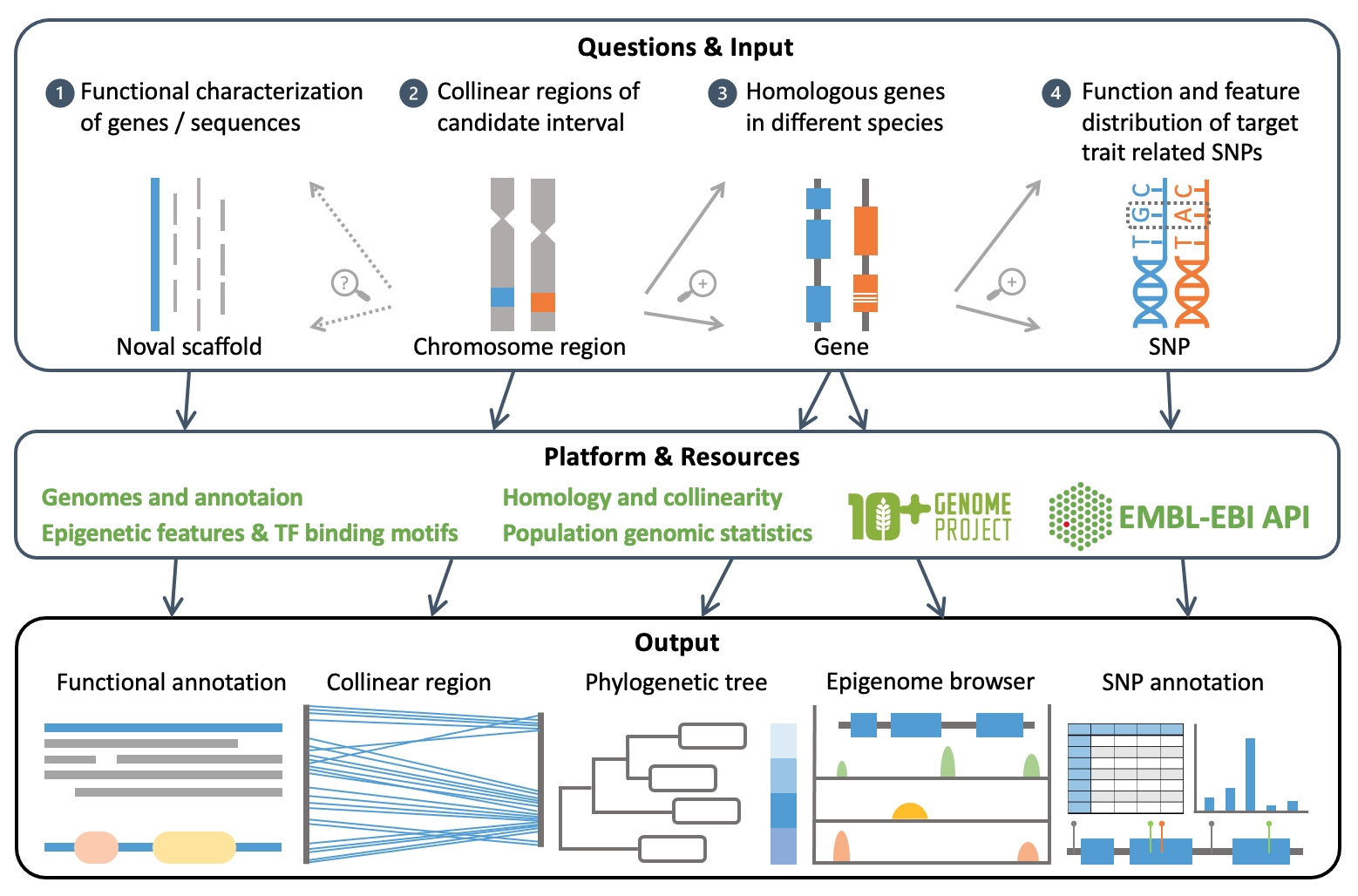
Triti-Map Annotation Platform is an online analysis module of Triti-Map. To locate causal variants and candidate genes or regulatory elements, Triti-Map integrated multi-omics data and various information from Triticeae species to provide a functional and evolutionary characterization of SNPs, genes, genomic regions, and new sequences related to the target trait.
The platform can perform various analyses, including SNP annotation and visual display, homologous gene analysis, collinearity analysis, and new sequence function annotation, providing richer reference information for Triticeae gene mapping.
You can also check out some of the FAQ you may encounter during use Triti-Map
Zhao F, Tian S, Li Z, et al. Utility of Triti-Map for bulk-segregated mapping of causal genes and regulatory elements in Triticeae[J]. Plant Communications, 2022: 100304.
https://doi.org/10.1016/j.xplc.2022.100304
Fei Zhao (zhaofei920810@gmail.com)
Lab Home Page:http://bioinfo.cemps.ac.cn/zhanglab/
Issues can be raised at: https://github.com/fei0810/Triti-Map/issues
We also encourage you to contribute to TriTi-Map! To fix bugs or add new features you need to create a Pull Request.
Fei Zhao (zhaofei920810@gmail.com)
Shilong Tian (tianshilong@cemps.ac.cn)
Thanks to @xuzhougeng who provided installation support for Bioconda, and thanks to @zwbao who offered Docker installation support.