modified based on https://github.com/FredHutch/crispr-screen-nf
Linux, MacOS, Windows(WSL)
clone the repository
git clone https://github.com/czbiohub/CRISPRflow.git
Go the repository directory, switch the branch if running branch other than master
cd CRISPRflow
Create conda environment and activate it
conda env create -f environment.yml
conda activate CRISPRflow
Pull docker images
bash helper_scripts/pull_docker_imgs.sh
Make the nextflow command executable
chmod a+x ./nextflow
Start the Docker program.
Check fastq files and create nextflow commands
conda activate CRISPRflow
python helper_scripts/check_files_and_get_nf_cmds.py --xlsx metadata/Naming_convention_example.xlsx
You should see the following output:

Start nextflow
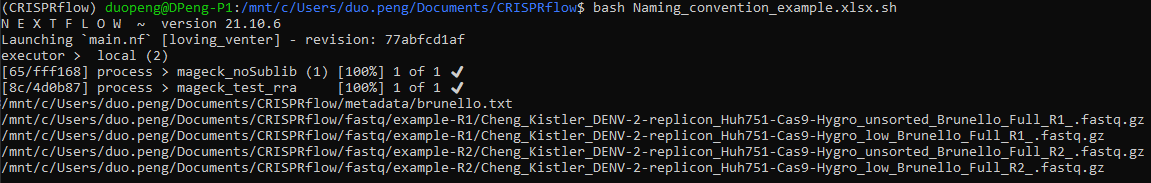
bash Naming_convention_example.xlsx.sh
You should see the following output:
- Fastq files
- A reference library file for each (sub)library
- A xlsx file that contains metadata and design of the analysis (example file:
metadata/Naming_convention_example.xlsx)
The metadata xlsx file will automatically generate path + names for the fastq.gz files, please make sure to move and rename your fastq.gz files accordingly
For file format, see MAGeCK manual: https://sourceforge.net/p/mageck/wiki/Home/
- Java issues (after installing java):
For most case, it can be fixed by explicitly specifying the version installed (v17 in the example)
unset JAVA_HOME
export JAVA_HOME=`/usr/libexec/java_home -v 17`
-
Error "executing process ... The command 'docker' could not be found ..."
You'll need to start the docker program on your computer -
Error "Both treatment and control are in the file name", but it's not the case
Captalize the first letter of your treatment names, for example:Infected,Uninfected