




MAESTRO(Model-based AnalysEs of Single-cell Transcriptome and RegulOme) is a comprehensive single-cell RNA-seq and ATAC-seq analysis suit built using snakemake. MAESTRO combines several dozen tools and packages to create an integrative pipeline, which enables scRNA-seq and scATAC-seq analysis from raw sequencing data (fastq files) all the way through alignment, quality control, cell filtering, normalization, unsupervised clustering, differential expression and peak calling, celltype annotation and transcription regulation analysis. Currently, MAESTRO support Smart-seq2, 10x-genomics, Drop-seq, SPLiT-seq for scRNA-seq protocols; microfudics-based, 10x-genomics and sci-ATAC-seq for scATAC-seq protocols.
- Release MAESTRO.
- Provide docker image for easy installation. Note, the docker does not include cellranger/cellranger ATAC, as well as the corresponding genome index. Please install cellranger/cellranger ATAC following the installation instructions.
- Fix some bugs and set LISA as the default method to predict transcription factors for scRNA-seq. Note, the docker includes the LISA conda environment, but does not include required pre-computed genome datasets. Please download hg38 or mm10 datasets and update the configuration following the installation instructions.
- Change the default alignment method of MAESTRO from cellranger to starsolo and minimap2 for accelerating the mapping time.
- Improve the memory efficiency of scATAC gene score calculation.
- Incorporate the installation of giggle into MAESTRO, and add web API for LISA function. All the core MAESTRO function can be installed through the conda environment now!
- Provide more documents for the QC parameters and add flexibility for other parameters in the workflow.
- Modify the regulatory potential model by removing the interfering peaks from adjacent genes and adjusting the weight of exon peaks. The "enhanced RP model" is set as the default gene activity scoring model with original "simple RP model" as a option.
- Modify the integration function of MAESTRO. The new function can output more intermediate figures and log files for diagnosing the possible failure in integrating rare populations.
- Add the function for annotating cell-types for scATAC-seq clusters based on public bulk chromatin accessibility data from Cistrome database (Note: Please update the giggle index to the latest).
- Provide the function of generating genome browser tracks at cluster level for scATAC-seq dataset visualization.
- Support peak calling at the cluster level now!
- For scATAC, MAESTRO can support fastq, bam, fragments.tsv.gz as the input of the scATAC-seq workflow.
- For scATAC, MAESTRO provides an option for users to skip the cell-type annotation step in the pipeline, and an option to choose the strategy for cell-type annotation (
RP-basedandpeak-based). - Provide small test data for test scRNA-seq and scATAC-seq pipeline (sampling from 10x fastq files).
- Add parameter validation before initializing the pipeline and provide more gracious error messages.
- Update R in MAESTRO conda package from 3.6.3 to 4.0.2, and Seurat from 3.1.2 to 3.1.5.
- Bug fixes (placeholder for v1.2.2 formal release)
- Update LISA to LISA2 which extends the original, runs faster, reduces dependencies, and adds useful CLI functions for pipeline integration. Please download the LISA2 data from human and mouse.
- Update conda dependencies to only requesting lowest versions.
- Fix the bugs in conda package installation channel.
- Update markers in the mouse.brain.ALLEN cell signature file.
- Fix the bugs to support the 10X .h5 file as the input format of MAESTRO scatac-genescore.
- Rename 'Adjusted RP model' to 'Enhanced RP model'.
- Fix the bugs in Snakefile to meet the latest version of snakemake.
- Update STAR reference indexes files for STAR -version 2.7.6a. Provide pre-built indexes for human and mouse.
- scATAC-seq multi-sample pipeline enabled. Deduplication can be set at cell or bulk level.
- Peak count matrix can be generated either on binary or raw count.
- LISA2 data will only be configured once in a given environment.
- Update web links for downloading reference data.
- Fix the bug in raw peak count matrix generation.
- Move from TravisCI to GitHub Actions for building package.
- LISA2 upgrades to v2.2.2. New LISA data are required for human and mouse. Please download to your computer and provide the path when initiating.
- Add LISA path as a variable in TF annotating function.
- Reduce the time and memory usage in the peak counting step.
- Fix the bug in the simple RP model for gene score calculation.
- Fix the bug in scATAC-Seq Snakefile.
- Linux/Unix
- Python (>= 3.7) for MAESTRO snakemake workflow
- R (>= 4.0.2) for MAESTRO R package
There are two ways to install MAESTRO -- to install the full workflow through Anaconda cloud; or to install only the R codes for exploring the processed data.
MAESTRO uses the Miniconda3 package management system to harmonize all of the software packages. Users can install the full solution of MAESTRO using the conda environment.
Use the following commands to install Minicoda3:
$ wget https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh
$ bash Miniconda3-latest-Linux-x86_64.shAnd then users can create an isolated environment for MAESTRO and install through the following commands:
$ conda config --add channels defaults
$ conda config --add channels liulab-dfci
$ conda config --add channels bioconda
$ conda config --add channels conda-forge
# To make the installation faster, we recommend using mamba
$ conda install mamba -c conda-forge
$ mamba create -n MAESTRO maestro=1.3.2 -c liulab-dfci
# Activate the environment
$ conda activate MAESTROIf users already have the processed datasets, like cell by gene or cell by peak matrix generate by Cell Ranger. Users can install the MAESTRO R package alone to perform the analysis from processed datasets.
$ R
> library(devtools)
> install_github("liulab-dfci/MAESTRO")The full MAESTRO workflow requires extra annotation files and tools:
-
MAESTRO depends on starsolo and minimap2 for mapping scRNA-seq and scATAC-seq dataset. Users need to generate the reference files for the alignment software and specify the path of the annotations to MAESTRO through command line options.
-
MAESTRO utilizes LISA2 to evaluate the enrichment of transcription factors based on the marker genes from scRNA-seq clusters. If users want to use LISA2, they need to download and install reference data either for human or for mouse locally and build the data according to the LISA2 document. The input gene set can be constituted of only official gene symbols, only RefSeq ids, only Ensembl ids, only Entrez ids, or a mixture of these identifiers.
-
MAESTRO utilizes giggle to identify enrichment of transcription factor peaks in scATAC-seq cluster-specific peaks. By default giggle is installed in MAESTRO environment. The giggle index for Cistrome database can be downloaded here (Note: Before v1.2.0, the giggle index
giggle.tar.gzcan be downloaded from http://cistrome.org/~galib/giggle.tar.gz. Since v1.2.0, please download the latest index giggle.all.tar.gz). Users need to download the file and provide the location of the giggle annotation to MAESTRO when using the ATACAnnotateTranscriptionFactor function.
usage: MAESTRO [-h] [-v]
{scrna-init,scatac-init,integrate-init, multi-scatac-init, samples-init, mtx-to-h5,count-to-h5,merge-h5,scrna-qc,scatac-qc,scatac-peakcount,scatac-genescore}
There are ten functions available in MAESTRO serving as sub-commands.
| Subcommand | Description |
|---|---|
scrna-init |
Initialize the MAESTRO scRNA-seq workflow. |
scatac-init |
Initialize the MAESTRO scATAC-seq workflow. |
integrate-init |
Initialize the MAESTRO integration workflow. |
multi-scatac-init |
Initialize the MAESTRO multi-sample scATAC-seq workflow. |
samples-init |
Initialize samples.json file in the current directory. |
mtx-to-h5 |
Convert 10X mtx format matrix to HDF5 format. |
count-to-h5 |
Convert plain text count table to HDF5 format. |
merge-h5 |
Merge multiple HDF5 files, e.g. different replicates. |
scrna-qc |
Perform quality control for scRNA-seq gene-cell count matrix. |
scatac-qc |
Perform quality control for scATAC-seq peak-cell count matrix. |
scatac-peakcount |
Generate peak-cell binary count matrix. |
scatac-genescore |
Calculate gene score based on the binarized scATAC peak count. |
Example of running MAESTRO can be found at the following galleries. Please use MAESTRO COMMAND -h to see the detail description for each option of each module.

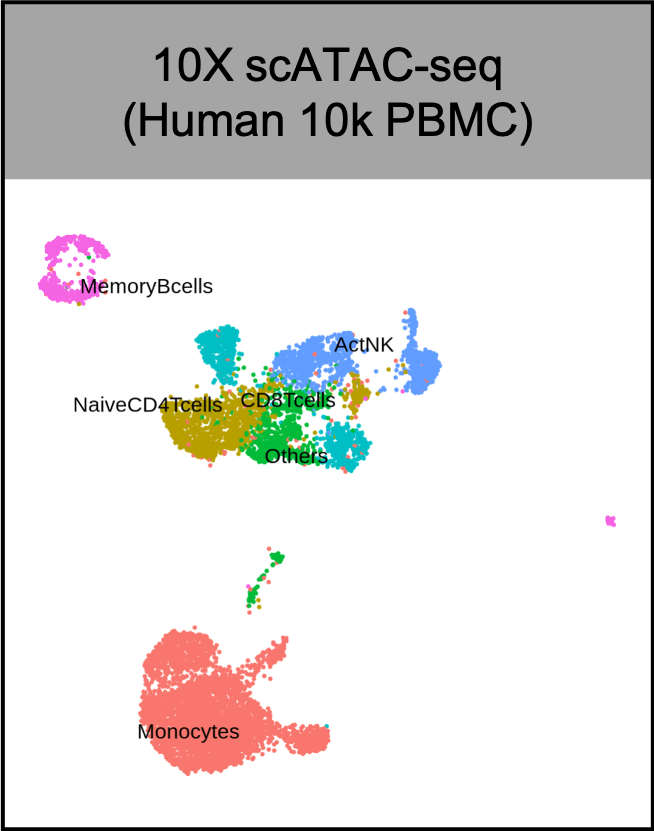

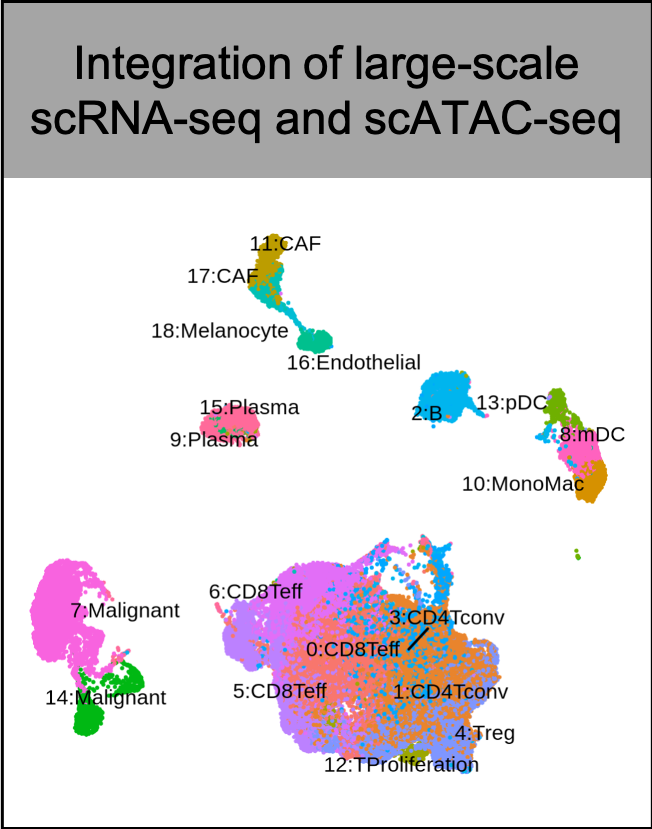
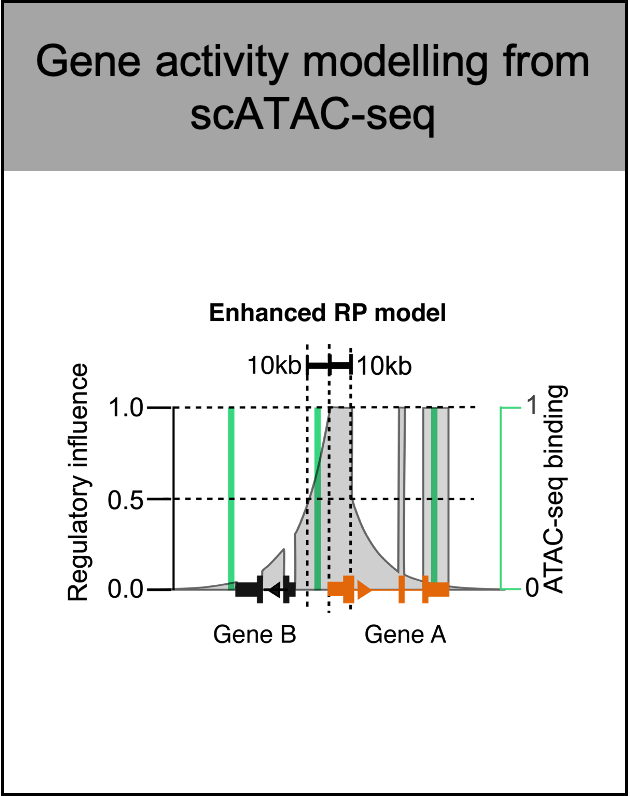

Wang C, Sun D, Huang X, Wan C, Li Z, Han Y, Qin Q, Fan J, Qiu X, Xie Y, Meyer CA, Brown M, Tang M, Long H, Liu T, Liu XS. Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol. 2020 Aug 7;21(1):198. doi: 10.1186/s13059-020-02116-x. PMID: 32767996; PMCID: PMC7412809.