tigeR is an R package designed for exploring biomarkers and constructing predictive models for immunotherapy response via built-in or custom immunotherapy gene expression data.
-
Built-in datasets: 1060 samples with immunotherapy clinical information from 11 melanoma datasets, 3 lung cancer datasets, 2 kidney cancer datasets, 1 gastric cancer dataset, 1 low-grade glioma dataset, 1 glioblastoma dataset and 1 head and neck squamous cell cancer dataset (all organized into R language ‘SummarizedExperiment’ objects).
-
23 immunotherapy response-related biomarkers from literature, multiple methods for analysis and visualization.
-
10 open source tumor microenvironment deconvolution methods including CIBERSORT, TIMER, ESTIMATE, IPS, xCell, EPIC, ConsensusTME, ABIS, quanTIseq, and MCPCounter. Several downstream method for analysis and visualization.
-
7 machine learning method for multi-modal prediction model construction and testing.

Overall design of tigeR
packages <- c("BiocManager", "devtools", "ggplot2", "pROC", "RobustRankAggreg")
for (package in packages) {
if (!require(package, character.only = TRUE)) {
install.packages(package)
}
}
devtools::install_github("YuLab-SMU/tigeR")
The workflow of tigeR is below, see more details in tigeR documentation.
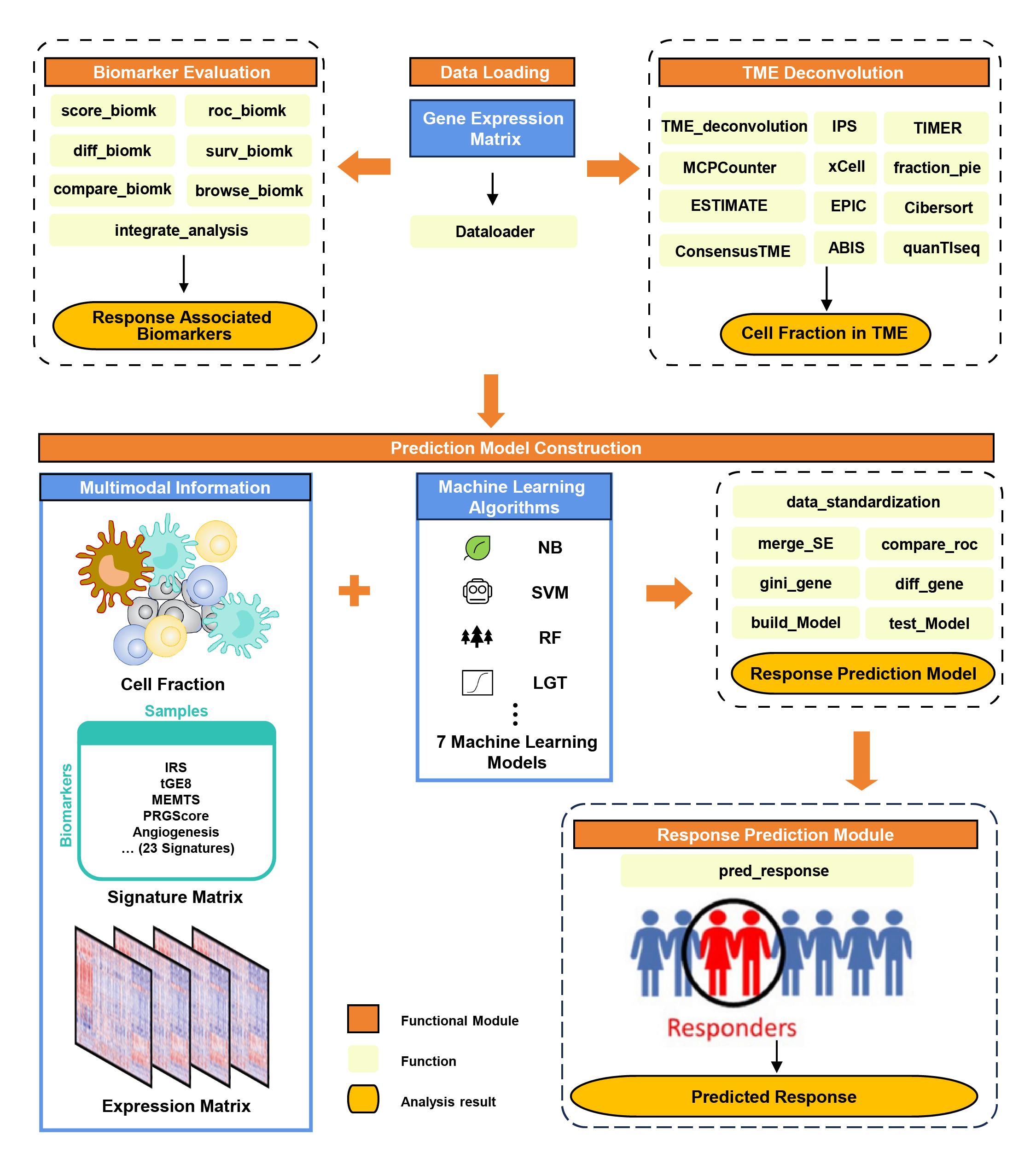
Workflow of tigeR