Tiny DFT is a minimalistic atomic Density Functional Theory (DFT) code, mainly for educational purposes. It only supports spherically symmetric atoms and local exchange-correlation functionals (at the moment only Dirac exchange).
The code is designed with the following criteria in mind:
-
It depends only on established scientific Python libraries: numpy, scipy, matplotlib and (the lesser known) autograd. The latter is a library for algorithmic differentiation, used to computed the analytic exchange(-correlation) potential and the grid transformation.
-
The numerical integration and differentiation algorithms should be precise enough to at least 6 significant digits for the total energy, but in many cases the numerical precision is better. Some integrals over Gaussian basis functions are computed analytically. The pseudo-spectral method with Legendre polynomials is used for the Poisson solver.
-
The total number of lines should be minimal and the source-code should be easy to understand, provided some background in DFT and spectral methods. As in most atomic DFT codes, the occupation numbers of the orbitals are all given the same value within one pair of angular and principal quantum number, to obtain a spherically symmetric density. The code only keeps track of the total number of electrons for each pair of quantum numbers.
-
Download the Tiny DFT repository. This can be done with your browser, after which you unpack the archive: https://github.com/theochem/tinydft/archive/master.zip. Or you can use git:
bash git clone https://github.com/theochem/tinydft.git cd tinydft -
Install the
tinydftpackage from source in development mode (for easy hacking):bash pip install -e .
To run a series of atomic DFT calculation, up to argon, run the mendelejev demo
bash tinydft-demo-mendelejev
This generates some screen output with SCF convergence, energy contributions and
the figures rho_z0*.png with the radial electron densities on a semi-log plot.
To modify the settings for these calculation, you can directly edit the source code.
When you make serious modifications to Tiny DFT, you can run the unit tests to make sure the original features still work.
For this, you first need to install pytest:
bash python3 -m pip install pytest pytest-cov pytest-regressions pandas --upgrade pytest
In order of increasing difficulty:
-
Change
tinydft/demo_mendelejev.pyto also make plots of the radial probability density, the occupied orbitals, the potentials (external, Kohn-Sham) with horizontal lines for the (higher but still negative) energy levels. Does this code reproduce the Rydberg spectral series well? (See https://en.wikipedia.org/wiki/Hydrogen_spectral_series#Rydberg_formula) -
Add a second unit test for the Poisson solver in
tests/test_tinydft.py. The current unit test checks if the Poisson solver can correctly compute the electrostatic potential of a spherical exponential density distribution (Slater-type). Add a similar test for a Gaussian density distribution. Use theerffunction fromscipy.specialto compute the analytic result. -
At the moment, the DFT calculations assume spin-unpolarized densities, which mainly affects the exchange-correlation energy. Change the code to work with spin-polarized densities. For this, also the occupation numbers for each angular momentum and principal quantum number should be split into a spin-up and spin-down occupation number.
-
Change the external potential to take into account the finite extent of the nucleus. You can use a Gaussian density distribution. Write a python script that combines isotope abundancies from NUBASE2016 and nucleotide radii from ADNDT to build a table of abundance-averaged nuclear radii.
-
Add a correlation energy density to the function
excfunctionand check if it improve the results in assignment (2). The following correlation functional has a good compromise between simplicity and accuracy: -
The provided implementation has a rigid algorithm to assign occupation numbers using the Klechkowski rule. Replace this by an algorithm that just looks for all the relevant lowest-energy orbitals at every SCF iteration.
-
Implement an SCF convergence test, which checks if the new Fock operator, in the basis of occupied orbitals from a previous iteration, is diagonal with orbital energies from the previous iteration on the diagonal
-
Implement the zeroth-order regular approximation to the Dirac equation (ZORA). ZORA needs a pro-atomic Kohn-Sham potential as input, which remains fixed during the SCF cycle. Add an outer loop where the first iteration is without ZORA and subsequent iterations use the Kohn-Sham potential from the previous SCF loop as pro-density for ZORA. (To avoid that the density diverges at the nucleus, assignment 4 should be implemented first.)
In ZORA, the following operator should be added to the Hamiltonian:
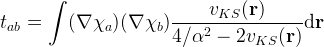
where the first factors are the gradients of the basis functions (similar to the kinetic energy operator). The Kohn-Sham potential from the previous outer iteration can be used. The parameter alpha is the dimensionless inverse fine-structure constant, see https://physics.nist.gov/cgi-bin/cuu/Value?alphinv and https://docs.scipy.org/doc/scipy/reference/constants.html (
inverse fine-structure constant). Before ZORA can be implemented, the formula needs to be worked out in spherical coordinates, separating it in a radial and an angular contribution. -
Extend the program to perform unrestricted Spin-polarized KS-DFT calculations. (Assignment 6 should done prior to this one.) In addition to the Aufbau rule, you now also have to implement the Hund rule. You also need to keep track of spin-up and spin-down orbitals. The original code uses the angular momentum quantum number,
angqnas keys in theeps_orbs_udictionary. Instead, you can now use(angqn, spinqn)keys. -
Extend the program to support (fractional) Hartree-Fock exchange.
-
Extend the program to support (meta) generalized gradient functionals.

The variable names are not always the shortest possible, e.g. atnum instead
of z, to make them more self-explaining and to comply with good practices.
alphas: Gaussian exponents in basis functionsatcharge: Atomic chargeatnum: Atomic numberangqn: Angular momentum (or azimuthal) quantum numbercoeffs: Expansion coefficients of a function in Gaussian primitives or Legendre polynomials.econf: Electronic configurationenergy_hartree: Hartree energy, i.e. classical electron-electron repulsion.eps: Orbital energieseps_orbs_u: A list of tuples of (orbital energy, orbital coefficients). One tuple for each angular momentum quantum number. The orbital coefficients represent the radial solutions U = R/r.energy_xc: Exchange-correlation energyexc: Exchange-correlation energy densityevals: Eigenvaluesevecs: Eigenvectorsext: Integrals for interaction with the external field (proton)fnvals: Function values on grid pointsfock: Fock operatoriscf: Current SCF iterationjxc: Hartree-Exchange-Correlation operatorkin_rad: Radial kinetic energy integralskin_ang: Angular kinetic energy integralsmaxangqn: Maximum angular quantum number of the occupied orbitalsnbasis: Number of basis functionsnelec: Number of electronsnscf: Number of SCF iterationsoccups: Occupation numbersolp: Overlap integralsorb_u: Orbital divided by r: U = R/rorb_r: Orbital: R = U*rpriqn: Primary quantum numbersrho: Electron density on grid pointsvhartree: Hartree potential, i.e. minus classical electrostatic potential due to the electrons.vol: Volume element in spherical coordinatesvxc: Exchange-correlation potential