This repository aims to provide a quick start guide to the installation, running, troubleshooting and tips of the Nextstrain workflow (https://nextstrain.org)
Example of Nextstrain output:
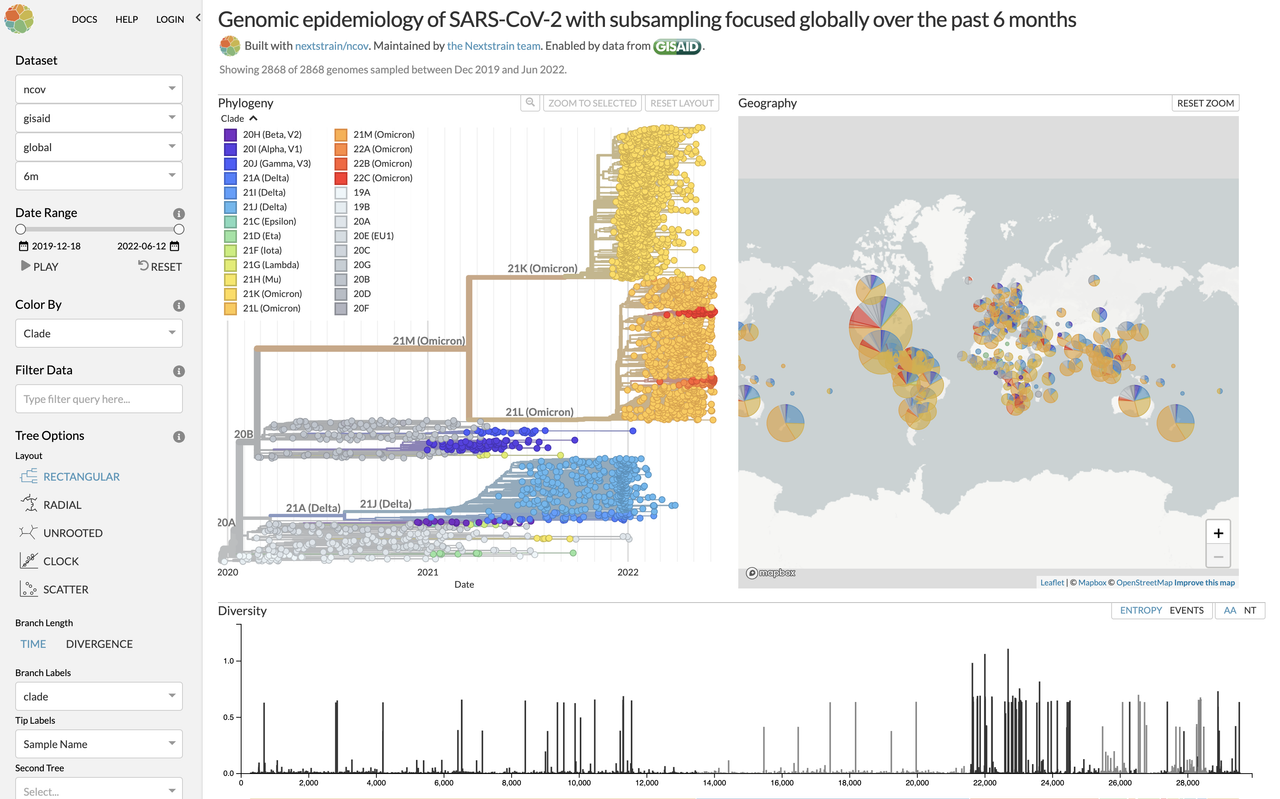
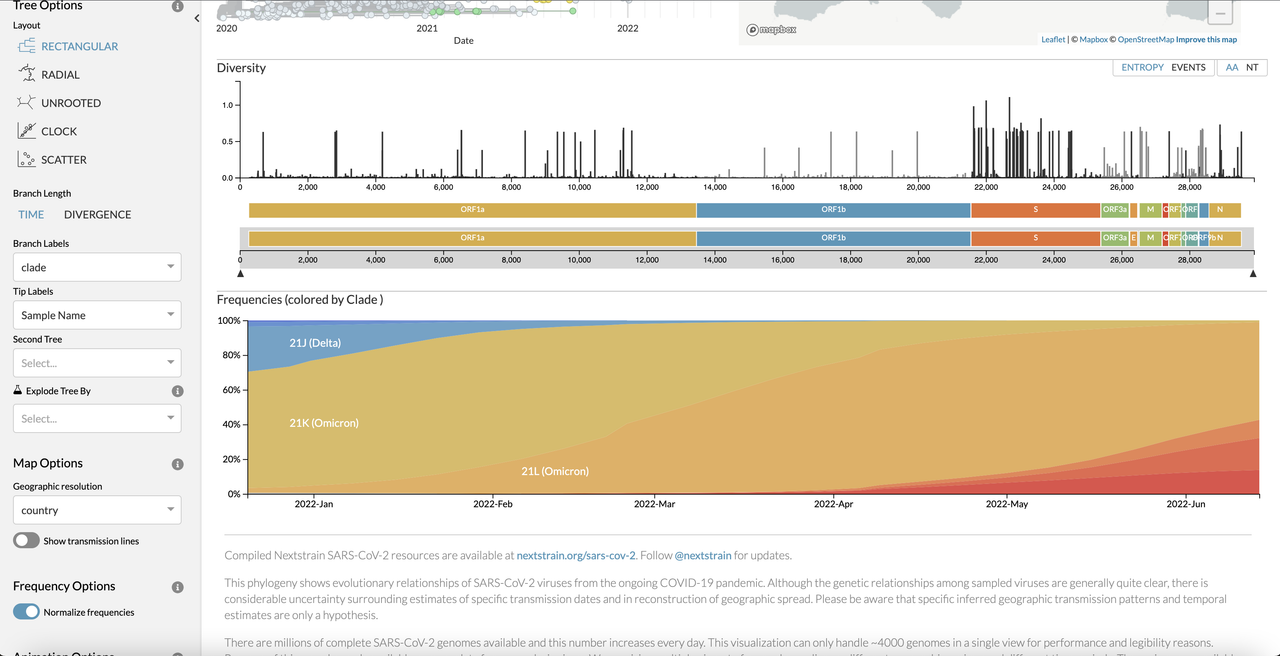
- Nextstrain is compatible with macOS, windows, WSL on windows, Ubuntu Linux and docker.
- Nextstrain runs on your own local environment, your data will not be published. (Unless its on GISAID, of course 😄 )
- The installation steps here will be for Ubuntu linux Native environment , if you use macOS you will find a file named macOS_installation_code.txt attached. Or if you use other OS system, you will find a link in the references section.
- In this quick guid we will take the SARS-CoV2 (ncov)* workflow as an example. However, Nextstrain provide workflows for multiple pathogens.
- The guide for preparing data mentioned here is for your local data.
- If you want to choose between Anaconda and miniconda, This link might help
- Anaconda installation guide
- Miniconda Installation guide
$ conda install -n base -c conda-forge mamba --yes
$ conda activate base
- Create a conda environment named nextstrain and install all the necessary software using mamba:
$ mamba create -n nextstrain -c conda-forge -c bioconda nextstrain-cli augur auspice nextalign snakemake git --yes
- Activate the conda environment:
$ conda activate mextstrain
- Confirm that the installation worked.
$ nextstrain check-setup --set-default
- The final output from the last command should look like this
Setting default environment to <option>., where<option>is the option chosen in the previous step.
Here you will find the list of pathogens that has a workflow in Nextstrain. https://nextstrain.org/pathogens
- If using the native runtime, install these additional packages necessary to run the ncovworkflow. Make sure to activate the correct conda environment.
$ conda activate nextstrain
$ mamba install -c conda-forge -c bioconda epiweeks nextclade nextalign pangolin pangolearn
Sometimes using conda instead of mamba works !
- Download the ncov workflow:
- Use
Gitto download a copy of the ncov repository containing the workflow and the testing data.
- Use
$ git clone https://github.com/nextstrain/ncov.git
4. After installation make sure that you have the following directories/files in your #PATH/nextstrain/ncov directory:
- snakefile; nextstrain use snakemake as their workflow managment software
- data; location of the final input data
- default; this contain all the defalt parameters for the workflow. Also, this directory contains two files you might need which are:
- lat_longs.tsv: if you want to add the latitude and longitude of regions or countries that is not included in the default. to be presented in the final map.
- color_schemes.tsv: you can take color scheme id numbers if you want to add colors to your additional regions or countries. (we will talk about this in the buid.yml preparation section)
- my_profile; includes example profiles and this is where you will have your run_directory which contain your data preparation files and the build.yml and config.yml
- results
- auspice: here is the visulization output
.jasonfiles location - workflow; contain workflow
- benchmark
- docs
- logs; you will find all the logs (also, helpful in case you face an error massage to identify the issue)
- narratives
- nextstrain_profiles
- scripts
#PATH: here insert your path
$ cd #PATH/nextstrain/ncov
$ conda activate nextstrain
$ nextstrain build . --cores all --configfile ncov-tutorial/example-data.yaml
Visualize test data results:
$ nextstrain view auspice/
-
you have more than one option:
- You want to analyze your own local data only
- you want to analyze your data with globally published data (GISAID)
- you want to analyze globally publised data only full GISAID database
-
before running any new run, go the the following directories (logs, auspec, and results) and move any previous outputs to a new directory (previous_runName) so the results won't overlap
-
go to /ncov/my_profiles and create a directory for the run
$ mkdir runName
$ cd runName
- copy the following files to your runName directory
$ cp ../example/builds.yaml ../example/config.yaml ../example/my_description.md . && touch colors.tsv
- choose the sequences data you want to include
- collect all the sequences into one fasta
$ cat *.new.fa > all.fasta
$ for i in `ls all.fasta`;do sed -i 's/"//' $i;done
- add the reference sequence to the all.fasta (Wuhan/Hu-1/2019)
- capitalize all the letters:
$ awk 'BEGIN{FS=" "}{if(!/>/){print toupper($0)}else{print $1}}' sequences.fasta > output.fasta
- make the sequences one line
$ awk '{if(NR==1) {print $0} else {if($0 ~ /^>/) {print "\n"$0} else {printf $0}}}' sequences.fasta > output.fasta
- make sure that the sequences header strings
>dose not contain any()[]{}|#><charecters.
-
create runName.metadata.tsv, you can edit the file via commandline or by excel or other editors. if you use excel or any editor to edit your file make sure that when you finish use this command to remove any foreign characters
$ sed -i 's/^M$//' runName.metadata.tsv -
this is the metadata columns structure: (Here we added the data for SARS-CoV2 reference genome as an example.)
Virus name virus gisaid_epi_isl genbank_accession date region country division location region_exposure country_exposure division_exposure segment length host age sex Nextstrain_clade pangolin_lineage GISAID_clade originating_lab submitting_lab authors url title paper_url date_submitted purpose_of_sequencing Wuhan/Hu-1/2019 betacoronavirus EPI_ISL_402125 ? 03-12-2020 Asia China China China Asia China ? genome 29903 unknown unknown ? B L National Institute for Communicable Disease Control and Prevention (ICDC) Chinese Center for Disease Control and Prevention (China CDC) National Institute for Communicable Disease Control and Prevention (ICDC) Chinese Center for Disease Control and Prevention (China CDC) Zhang,Y.-Z., Wu,F., Chen,Y.-M., Pei,Y.-Y., Xu,L., Wang,W., Zhao,S., Yu,B., Hu,Y., Tao,Z.-W., Song,Z.-G., Tian,J.-H., Zhang,Y.-L., Liu,Y., Zheng,J.-J., Dai,F.-H., Wang,Q.-M., She,J.-L. and Zhu,T.-Y. https://www.gisaid.org/ ? ? 30-12-2020 ? ? - date format must be as: YYYY-MM-DD
prepare the builds and config files based on what you want to do in this run. for example the basic things you need to run the pipeline is the following: if you want to do other things, you will find a reference in the reference section that might help.
$ vi builds.yaml
inputs:
- name: runName
metadata: data/runName.metadata.tsv
sequences: data/runName.sequences.fasta
# the files below is not mandatory, if you have it and want to use just remove the # sign
#files:
#colors: "my_profiles/runName/colors.tsv"
#description: "my_profiles/runName/description.md"
$ vi config.yaml
configfile:
- defaults/parameters.yaml # Pull in the default values
- my_profiles/runName/builds.yaml
# Set the maximum number of cores you want Snakemake to use for this pipeline.
cores: 1
# Always print the commands that will be run to the screen for debugging.
printshellcmds: True
# Always print log files of failed jobs to the screen.
show-failed-logs: True
- it is always useful to write down a small discreption on your run and what you are aiming to do get from this dataset in the
my_description.mdfile. (this step is unnecessary) 🧐
- The files you prepare:
- genome sequences .fasta file (runName.sequences.fasta)
- A corresponding TSV file with metadata describing each sequence (runName.metadata.tsv)
- configration file for the workflow that says how to use those data. (config.yml)
- build.yml file to locate the path of the data and other informations
- we need to provide references sequences which is really important for reading the phylogenetic time tree. This reference sequence must be included in the (runName.sequences.fasta) and its data in (runName.metadata.tsv)
- Move the following files:
$ cp runName.sequences.fasta #PATH/nextstrain/ncov/data
$ cp runName.metadata.tsv #PATH/nextstrain/ncov/data
(base) $ conda activate nextstrain
(nextstrain) $ snakemake --profile my_profiles/runName/ -p
- when your run is finished, and no error massages appeared. go to the
#PATH/nextstrain/ncov/auspice - open this link https://auspice.us/
- drag the runName.json file and drop it in the https://auspice.us/ to visualize your data. (you can find the example data attached)
Nextstrain have created a website hub (Nextstrain discussion) that you can visit to seek help with anything that you might face, or to just have an insight. They have been of great help
https://docs.nextstrain.org/projects/cli/en/stable/commands/update/
- nextstrain/ncov
- changes logs
- A Getting Started Guide to the Genomic Epidemiology of SARS-CoV-2
- Data preparation guide
- Prepare your own local data
- SARS-CoV-2 clade naming strategy for 2022
- Data Sharing via GitHub
- Nextstrain CLI
Here we share some of the error faced and how to solve them 🤓 👌
- error:
Error in rule calculate_epiweeks:
https://www.roseindia.net/answers/viewqa/pythonquestions/95199-ModuleNotFoundError-No-module-named-epi-weeks.html
jobid: 30
output: results/default-build/epiweeks.json
log: logs/calculate_epiweeks_default-build.txt (check log file(s) for error message)
shell:
python3 scripts/calculate_epiweek.py --metadata results/default-build/metadata_adjusted.tsv.xz --output-node-data results/default-build/epiweeks.json 2>&1 | tee logs/calculate_epiweeks_default-build.txt
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Logfile logs/calculate_epiweeks_default-build.txt:
Traceback (most recent call last):
File "scripts/calculate_epiweek.py", line 4, in <module>
import epiweeks
ModuleNotFoundError: No module named 'epiweeks'
- Solution:
$ conda install -c bioconda epiweeks
- error:
Error in rule refine:
jobid: 5
output: results/default-build/tree.nwk, results/default-build/branch_lengths.json
log: logs/refine_default-build.txt (check log file(s) for error message)
shell:
augur refine --tree results/default-build/tree_raw.nwk --alignment results/default-build/aligned.fasta --metadata results/default-build/metadata_adjusted.tsv.xz --output-tree results/default-build/tree.nwk --output-node- data results/default-build/branch_lengths.json --root Wuhan/Hu-1/2019 --timetree --clock-rate 0.0008 --clock-std-dev 0.0004 --coalescent opt --date-inference marginal --divergence-unit mutations --date-confidence --no-covariance --clock-filter-iqd 4 2>&1 | tee logs/refine_default-build.txt
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Logfile logs/refine_default-build.txt:
augur refine is using TreeTime version 0.8.4
1.67 TreeTime.reroot: with method or node: Wuhan/Hu-1/2019
Traceback (most recent call last):
File "/home/bioinformatics/miniconda3/envs/nextstrain/bin/augur", line 10, in <module>
sys.exit(main())
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/augur/__main__.py", line 10, in main
return augur.run( argv[1:] )
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/augur/__init__.py", line 75, in run
return args.__command__.run(args)
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/augur/refine.py", line 206, in run
tt = refine(tree=T, aln=aln, ref=ref, dates=dates, confidence=args.date_confidence,
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/augur/refine.py", line 42, in refine
tt.clock_filter(reroot=reroot, n_iqd=clock_filter_iqd, plot=False) #use whatever was specified
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/treetime/treetime.py", line 345, in clock_filter
self.reroot(root='least-squares' if reroot=='best' else reroot, covariation=False, clock_rate=fixed_clock_rate)
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/treetime/treetime.py", line 471, in reroot
raise UnknownMethodError('TreeTime.reroot -- ERROR: unsupported rooting mechanisms or root not found')
treetime.UnknownMethodError: TreeTime.reroot -- ERROR: unsupported rooting mechanisms or root not found
-
Solution: https://discussion.nextstrain.org/t/unsupporting-rooting-mechanisms-or-root-not-found/568
-
error:
Error in rule recency:
jobid: 23
output: results/default-build/recency.json
log: logs/recency_default-build.txt (check log file(s) for error message)
shell:
python3 scripts/construct-recency-from-submission-date.py --metadata results/default-build/metadata_adjusted.tsv.xz --output results/default-build/recency.json 2>&1 | tee logs/recency_default-build.txt
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Logfile logs/recency_default-build.txt:
Traceback (most recent call last):
File "scripts/construct-recency-from-submission-date.py", line 41, in <module>
node_data['nodes'][strain] = {'recency': get_recency(d['date_submitted'], ref_date)}
File "scripts/construct-recency-from-submission-date.py", line 7, in get_recency
date_submitted = datetime.strptime(date_str, '%Y-%m-%d').toordinal()
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/_strptime.py", line 568, in _strptime_datetime
tt, fraction, gmtoff_fraction = _strptime(data_string, format)
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/_strptime.py", line 349, in _strptime
raise ValueError("time data %r does not match format %r" %
ValueError: time data '1/12/2020' does not match format '%Y-%m-%d'
-
Solution: change the date format in the metadata.tsv
-
error:
Error in rule sanitize_metadata:
jobid: 12
output: results/sanitized_metadata_run.tsv.xz
log: logs/sanitize_metadata_run.txt (check log file(s) for error message)
shell:
python3 scripts/sanitize_metadata.py --metadata data/gisaid_N501TGR_KSA.metadata.tsv --metadata-id-columns strain name 'Virus name' --database-id-columns 'Accession ID' gisaid_epi_isl genbank_accession --parse-location-field Location --rename-fields 'Virus name=strain' Type=type 'Accession ID=gisaid_epi_isl' 'Collection date=date' 'Additional location information=additional_location_information' 'Sequence length=length' Host=host 'Patient age=patient_age' Gender=sex Clade=GISAID_clade 'Pango lineage=pango_lineage' pangolin_lineage=pango_lineage Lineage=pango_lineage 'Pangolin version=pangolin_version' Variant=variant 'AA Substitutions=aa_substitutions' aaSubstitutions=aa_substitutions 'Submission date=date_submitted' 'Is reference?=is_reference' 'Is complete?=is_complete' 'Is high coverage?=is_high_coverage' 'Is low coverage?=is_low_coverage' N-Content=n_content GC-Content=gc_content --strip-prefixes hCoV-19/ SARS-CoV-2/ --output results/sanitized_metadata_run.tsv.xz 2>&1 | tee logs/sanitize_metadata_run.txt
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Logfile logs/sanitize_metadata_run.txt:
Traceback (most recent call last):
File "scripts/sanitize_metadata.py", line 394, in <module>
metadata = filter_duplicates(
File "scripts/sanitize_metadata.py", line 313, in filter_duplicates
metadata = metadata.reset_index().merge(
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/pandas/core/frame.py", line 9190, in merge
return merge(
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/pandas/core/reshape/merge.py", line 106, in merge
op = _MergeOperation(
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/pandas/core/reshape/merge.py", line 709, in __init__
self._validate(validate)
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/pandas/core/reshape/merge.py", line 1425, in _validate
raise MergeError(
pandas.errors.MergeError: Merge keys are not unique in left dataset; not a one-to-one merge
-
Solution: change the first column header to Virus name
-
error:
Error in rule filter:
jobid: 9
output: results/filtered_nnn.fasta.xz
log: logs/filtered_nnn.txt (check log file(s) for error message)
shell:
augur filter --sequences results/masked_nnn.fasta.xz --metadata results/sanitized_metadata_nnn.tsv.xz --include defaults/include.txt --max-date 2021-11-08 --min-date 2019.74 --exclude-ambiguous-dates-by any --exclude defaults/exclude.txt results/to-exclude_nnn.txt --exclude-where division='USA' --min-length 27000 --output results/filtered_nnn.fasta 2>&1 | tee logs/filtered_nnn.txt;
xz -2 results/filtered_nnn.fasta
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Logfile logs/filtered_nnn.txt:
Note: You did not provide a sequence index, so Augur will generate one. You can generate your own index ahead of time with `augur index` and pass it with `augur filter --sequence-index`.
ERROR: All samples have been dropped! Check filter rules and metadata file format.
102 strains were dropped during filtering
51 had no metadata
51 had no sequence data
-
Solution: check that the string name matched between the metadata and the sequences.
-
error:
Error in rule filter:
jobid: 9
output: results/filtered_nnn.fasta.xz
log: logs/filtered_nnn.txt (check log file(s) for error message)
shell:
augur filter --sequences results/masked_nnn.fasta.xz --metadata results/sanitized_metadata_nnn.tsv.xz --include defaults/include.txt --max-date 2021-11-08 --min-date 2019.74 --exclude-ambiguous-dates-by any --exclude defaults/exclude.txt results/to-exclude_nnn.txt --exclude-where division='USA' --min-length 27000 --output results/filtered_nnn.fasta 2>&1 | tee logs/filtered_nnn.txt;
xz -2 results/filtered_nnn.fasta
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Logfile logs/filtered_nnn.txt:
Note: You did not provide a sequence index, so Augur will generate one. You can generate your own index ahead of time with `augur index` and pass it with `augur filter --sequence-index`.
ERROR: All samples have been dropped! Check filter rules and metadata file format.
51 strains were dropped during filtering
1 of these were dropped because of their ambiguous date in any
50 of these were dropped because of their date (or lack of date)
-
Solution: correct the date formats in the metadata
-
error:
Error in rule refine:
jobid: 5
output: results/default-build/tree.nwk, results/default-build/branch_lengths.json
log: logs/refine_default-build.txt (check log file(s) for error message)
shell:
augur refine --tree results/default-build/tree_raw.nwk --alignment results/default-build/aligned.fasta --metadata results/default-build/metadata_adjusted.tsv.xz --output-tree results/default-build/tree.nwk --output-node-data results/default-build/branch_lengths.json --root Wuhan/Hu-1/2019 --timetree --clock-rate 0.0008 --clock-std-dev 0.0004 --coalescent opt --date-inference marginal --divergence-unit mutations --date-confidence --no-covariance --clock-filter-iqd 4 2>&1 | tee logs/refine_default-build.txt
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Logfile logs/refine_default-build.txt:
augur refine is using TreeTime version 0.8.4
0.78 ***WARNING: TreeAnc._attach_sequences_to_nodes: NO SEQUENCE FOR LEAF:
nCov2020_03-21-nnn-000987_03-2021
Traceback (most recent call last):
File "/home/bioinformatics/miniconda3/envs/nextstrain/bin/augur", line 10, in <module>
sys.exit(main())
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/augur/__main__.py", line 10, in main
return augur.run( argv[1:] )
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/augur/__init__.py", line 75, in run
return args.__command__.run(args)
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/augur/refine.py", line 206, in run
tt = refine(tree=T, aln=aln, ref=ref, dates=dates, confidence=args.date_confidence,
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/augur/refine.py", line 36, in refine
tt = TreeTime(tree=tree, aln=aln, ref=ref, dates=dates,
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/treetime/treetime.py", line 34, in __init__
super(TreeTime, self).__init__(*args, **kwargs)
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/treetime/clock_tree.py", line 67, in __init__
super(ClockTree, self).__init__(*args, **kwargs)
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/treetime/treeanc.py", line 162, in __init__
self._check_alignment_tree_gtr_consistency()
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/treetime/treeanc.py", line 380, in _check_alignment_tree_gtr_consistency
raise MissingDataError("TreeAnc._check_alignment_tree_gtr_consistency: At least 30\\% terminal nodes cannot be assigned a sequence!\n"
treetime.MissingDataError: TreeAnc._check_alignment_tree_gtr_consistency: At least 30\% terminal nodes cannot be assigned a sequence!
Are you sure the alignment belongs to the tree?
-
Solution: 1- match the sequences string name with the strain colimn in the meatdata 2- try to drop the 17 sequences mentioned 3- try to replace the / with _
https://discussion.nextstrain.org/t/an-attributeerror-nonetype-object-has-no-attribute-shape-when-building-phylogenetic-tree-using-augur/401/2 https://discussion.nextstrain.org/t/error-unsupported-rooting-mechanisms-or-root-not-found/381 -
error:
Error in rule refine:
jobid: 5
output: results/default-build/tree.nwk, results/default-build/branch_lengths.json
log: logs/refine_default-build.txt (check log file(s) for error message)
shell:
augur refine --tree results/default-build/tree_raw.nwk --alignment results/default-build/aligned.fasta --metadata results/default-build/metadata_adjusted.tsv.xz --output-tree results/default-build/tree.nwk --output-node-data results/default-build/branch_lengths.json --root Wuhan/Hu-1/2019 --timetree --clock-rate 0.0008 --clock-std-dev 0.0004 --coalescent opt --date-inference marginal --divergence-unit mutations --date-confidence --no-covariance --clock-filter-iqd 4 2>&1 | tee logs/refine_default-build.txt
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Logfile logs/refine_default-build.txt:
augur refine is using TreeTime version 0.8.4
0.84 TreeTime.reroot: with method or node: Wuhan/Hu-1/2019
Traceback (most recent call last):
File "/home/bioinformatics/miniconda3/envs/nextstrain/bin/augur", line 10, in <module>
sys.exit(main())
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/augur/__main__.py", line 10, in main
return augur.run( argv[1:] )
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/augur/__init__.py", line 75, in run
return args.__command__.run(args)
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/augur/refine.py", line 206, in run
tt = refine(tree=T, aln=aln, ref=ref, dates=dates, confidence=args.date_confidence,
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/augur/refine.py", line 42, in refine
tt.clock_filter(reroot=reroot, n_iqd=clock_filter_iqd, plot=False) #use whatever was specified
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/treetime/treetime.py", line 345, in clock_filter
self.reroot(root='least-squares' if reroot=='best' else reroot, covariation=False, clock_rate=fixed_clock_rate)
File "/home/bioinformatics/miniconda3/envs/nextstrain/lib/python3.8/site-packages/treetime/treetime.py", line 471, in reroot
raise UnknownMethodError('TreeTime.reroot -- ERROR: unsupported rooting mechanisms or root not found')
treetime.UnknownMethodError: TreeTime.reroot -- ERROR: unsupported rooting mechanisms or root not found
Shutting down, this might take some time.
Exiting because a job execution failed. Look above for error message
-
solution: 1- there is an error with the alignemnt, check the log: align_nnn. try to change the fasta file from inverted to single line fasta:
$ awk '{if(NR==1) {print $0} else {if($0 ~ /^>/) {print "\n"$0} else {printf $0}}}' interleaved.fasta > singleline.fasta2- Wuhan/Hu-1/2019 not being part of the dataset: https://githubmemory.com/repo/nextstrain/augur/issues/744 in metadata: WuhAn/Hu-1/2019 in sequences: WuhAn/Hu-1/2019 change both to: Wuhan/Hu-1/2019 -
error:
Building DAG of jobs...
Error: Directory cannot be locked. Please make sure that no other Snakemake process is trying to create the same files in the following directory:
/mnt/e/Bioinformatics/nextstrain/nextstrain/ncov
If you are sure that no other instances of snakemake are running on this directory, the remaining lock was likely caused by a kill signal or a power loss. It can be removed with the --unlock argument.
- solution:
- make sure that no tasks running
$ htop- run this code , then run your run
$ (nextstrain) bioinformatics@nnnn:/mnt/e/Bioinformatics/nextstrain/nextstrain/ncov$ snakemake --profile my_profiles/nnn/012020-122021/ -p --unlock
- error:
Traceback (most recent call last):
File "/mnt/e/Bioinformatics/nextstrain/nextstrain/ncov/scripts/sanitize_metadata.py", line 2, in <module>
from augur.io import open_file, read_metadata
ModuleNotFoundError: No module named 'augur'
[Sun Apr 3 12:34:36 2022]
Error in rule sanitize_metadata:
jobid: 12
output: results/sanitized_metadata_nnn.tsv.xz
log: logs/sanitize_metadata_nnn.txt (check log file(s) for error message)
shell:
python3 scripts/sanitize_metadata.py --metadata data/nnn.metadata.tsv --metadata-id-columns strain name 'Virus name' --database-id-columns 'Accession ID' gisaid_epi_isl genbank_accession --parse-location-field Location --rename-fields 'Virus name=strain' Type=type 'Accession ID=gisaid_epi_isl' 'Collection date=date' 'Additional location information=additional_location_information' 'Sequence length=length' Host=host 'Patient age=patient_age' Gender=sex Clade=GISAID_clade 'Pango lineage=pango_lineage' pangolin_lineage=pango_lineage Lineage=pango_lineage 'Pangolin version=pangolin_version' Variant=variant 'AA Substitutions=aa_substitutions' aaSubstitutions=aa_substitutions 'Submission date=date_submitted' 'Is reference?=is_reference' 'Is complete?=is_complete' 'Is high coverage?=is_high_coverage' 'Is low coverage?=is_low_coverage' N-Content=n_content GC-Content=gc_content --strip-prefixes hCoV-19/ SARS-CoV-2/ --output results/sanitized_metadata_nnn.tsv.xz 2>&1 | tee logs/sanitize_metadata_nnn.txt
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Logfile logs/sanitize_metadata_nnn.txt:
Traceback (most recent call last):
File "/mnt/e/Bioinformatics/nextstrain/nextstrain/ncov/scripts/sanitize_metadata.py", line 2, in <module>
from augur.io import open_file, read_metadata
ModuleNotFoundError: No module named 'augur'
Shutting down, this might take some time.
Exiting because a job execution failed. Look above for error message
Complete log: /mnt/e/Bioinformatics/nextstrain/nextstrain/ncov/.snakemake/log/2022-04-03T123435.417174.snakemake.log
- solution:
- i deleted the entire nextstrain env, apperantly there is an update https://docs.nextstrain.org/projects/ncov/en/latest/reference/change_log.html nextstrain/ncov#476
- https://docs.nextstrain.org/en/latest/install.html#installation-steps
- https://docs.nextstrain.org/projects/ncov/en/latest/analysis/setup.html