##目录
- 写在前面
- 关于WAM
- 代码说明
- 尾巴
关于WAM模型,我本来想写三个版本的代码,c、php、java,最先拿php实现了模型的功能,但是代码写的不够漂亮,在这里还是没有提交到库上。后来花了差不多五天实现用c重写,代码量约为1K,实现了全部功能,现托管于github上,与大家交流共享交流,欢迎大家批评指正!
-
操作系统: Ubuntu Server 14.04 (Why linux,why not Windows? 我曾经也一直在问这个问题,现在我放弃纠结了...)
-
编译器:gcc(gcc编译器默认不在ubuntu的发行包中,需要手动安装,执行这个命令即可:
$ sudo apt-get install gcc) -
make编译(可选)
-
如何获取代码:
如果你了解git版本管理工具,可以在linux中直接使用git命令行获取代码,执行
$ git clone https://github.com/shaojunyu/WAM.git
即可。(git软件包默认不在ubuntu的发行包中,需要手动安装$ sudo apt-get install git)如果不喜欢git,也可以直接下载。在网页中点击
Download ZIP就可以了,![][2]会直接下载一个压缩包,解压之后就可以看到文件了,代码位于code文件夹中. [2]:https://raw.githubusercontent.com/shaojunyu/WAM/c/image/download.png -
文件目录结构,如下图所示。在
code文件件中,有7个代码文件,一个makefile编译规则文件,外加两个数据文件夹Testing和Training,分别用于存储测试数据文件和训练数据文件,可以修改代码,但请勿随意修改目录结构!![][1] [1]: https://raw.githubusercontent.com/shaojunyu/WAM/c/image/dir.png
-
编译 编译可以使用这两个编译命令(二选一)
$ gcc -o WAM_Bin main.c jwHash.c -lm
或
$ make(如果你的linux环境中有make编译器) -
运行
编译成功后,会在当前目录下生成WAN_Bin可执行文件, 输入命令$ ./WAM_Bin即可运行
运行结果如下图所示:
没查到关于这个模型没查到很多资料。。。上课的时候也没怎么听明白。。。课下和几个同学讨论了一下,说一下我们自己的理解。我们可以暂且脱离生物学问题,将其抽象为一个简单的字符串模式匹配的问题。首先,总体的流程图如下所示:
![][follow]
[follow]:https://raw.githubusercontent.com/shaojunyu/WAM/c/image/follow.png
在训练的数据集中,我们可以获得不同剪接位点附近短序列(在这里,我们以9bp长度为例,我的代码中也是这么实现的),然后对这些短序列中各个位置碱基出现的规律进行统计。
![][3]
[3]:https://raw.githubusercontent.com/shaojunyu/WAM/c/image/9.png
如图,我们对这些短序列进行编号。之后,我们主要统计出两个数据表,一个是前景概率表,一个是背景概率表。前景概率表利用简介位点附近的短序列计算得来,背景概率由序列文件中随机抽取的其他任意短序列计算得来。两个概率表中包含的数据是 短序列中位置1某种碱基出现的概率,之后的数据是条件概率,考虑前一位点碱基与该位点碱基的关联性。在代码中,利用哈希表来存储数据表,主要是考虑利用哈希表的键(KEY)来存储位置和该位点碱基的信息,利用值(VALUE)来存储概率值。如 '1a'=>'0.25' 这样一个键值对表示在第一位出现剪辑a的概率为0.25;'9at' => '0.01',这样一个键值对表示在第八位为a的情况下,第九位出现碱基t的概率为0.01。
得到前景概率表和背景概率表之后,就可以按照WAM的打分公式(如下图)对测试数据集中任意9个碱基长度的短序列进行打分。p+代表前景概率,p-代表背景概率
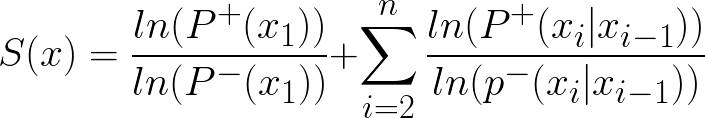
打完分分之后,我们可以规定一个阈值,认为分数在这个阈值之上的短序列,其第三个第四个碱基之间即为一个剪接位点。这样我们就可以得到预测的剪接位点,将预测的位点与真实的位点进行比对,获得Sn和Sp。调整分数的阈值,以提高Sn和Sp。
-
main.c:main函数所在地,程序的入口。实现了程序的主流程。我得说这个main函数写的很糟糕,竟然超过了200行代码,超过100行的main函数写的都不能算好的,main函数中还需要抽象出一些功能独立到单独的文件中 -
jwHash.hjwHash.c:这两个文件是利用C实现哈希表的,提供了操作哈希表的各宗API。这是我从github上找的开源项目,链接: Simple hash table implementation for C. -
file.h: 该文件中只有一个重要的函数file_to_short_seq, 用于读入存储序列的txt文档,并将其转化成短的序列存储到链表中 -
sequence.h: 定义了Short_seq链表结构体及其相关的操作函数,以及计算概率表的函数caculate_p_table,预测位点函数predict_seq -
queue.h: 定义了抽象的队列结构体,定义了对链表的操纵宏 -
其他具体问题可以参看代码中的注释或者咱们直接交流
C:
![][c]
[c]:https://pic1.zhimg.com/947c4dceaab030d4c9253e83fce040a8_b.jpg
Java:
![][java]
[java]:https://pic3.zhimg.com/61ec124e921ea95354ba5870365093d6_b.jpg
Python:
![][py]
[py]:https://pic2.zhimg.com/bbc915dbf9a893c04fae4795b24695c5_b.jpg
PHP:
![][php]
[php]:https://pic1.zhimg.com/96e0257002c80afdcc6b36ebb55b6f6c_b.jpg
Perl:
![][perl]
[perl]:https://pic2.zhimg.com/943bc5dd51e2c487193c1dfaeca3ea81_b.jpg
C++:
![][c++]
[c++]:https://pic1.zhimg.com/ab40d9b2260e1879a67e8e9ffc3f9dd8_b.jpg
Java: 我是面向对象的企业级开发语言
C: 我比你快...
Python: 我是面向对象的脚本语言
C: 我比你快...
PHP: 我是世界上最好的语言 ^-^
C: 我比你快...
Perl: 我是文本处理小能手
C: 我比你快...
C++: ...
C: ...
你遇到了一群敌人!
你要怎么办呢?
C:拿出一根棍子,一个一个把敌人砸死。
C++:用机关枪。
Java:我打电话叫十万个基佬!
Perl:我会功夫!
Python:用氢弹。
PHP:让我们把敌人老死吧!
开个玩笑,除非想不开,否则千万不要轻易尝试用c去写类似这样主要以文本处理为主的程序。没有面向对象,要用结构体来构造复杂的数据结构;没有字符串类型,要用字符指针逐个字节的处理;没有可变数组,要用自己实现链表来动态扩展数组;没有哈希表,要自己动手DIY;没有垃圾内存回收机制,要跪着把自己申请的内存一点点释放;整个项目的c代码量约为1k,但是其中只有约100行的核心逻辑代码,其他的有500行左右是去实现哈希表的,有50行左右是实现链表,还有几百行的文件读入及处理的代码。这些工作如果换成脚本语言来写的话,最多不会超过两百行代码,我们也可以把更多的精力放在业务逻辑本身上,不需要花太多的时间去给自己造工具!推荐大家使用Python或Perl
Acknowledgements: Yang Xu, Zhenyu Zhang, Hongjun Li