ProtSSN: Semantical and Topological Protein Encoding Toward Enhanced Bioactivity and Thermostability
- [2024.4.28] Our paper is under review on eLife, 10.7554/eLife.98033.
- [2023.12.23] Our ensemble ProtSSN achieves a spearman score of 0.449 on ProteinGym v1.0. You can view and compare different baseline models on the ProteinGym website.
Fusion of protein sequence and structural information, using denoising pre-training network for protein engineering (zero-shot). Please note that we do not require MSA or evolutionary information.
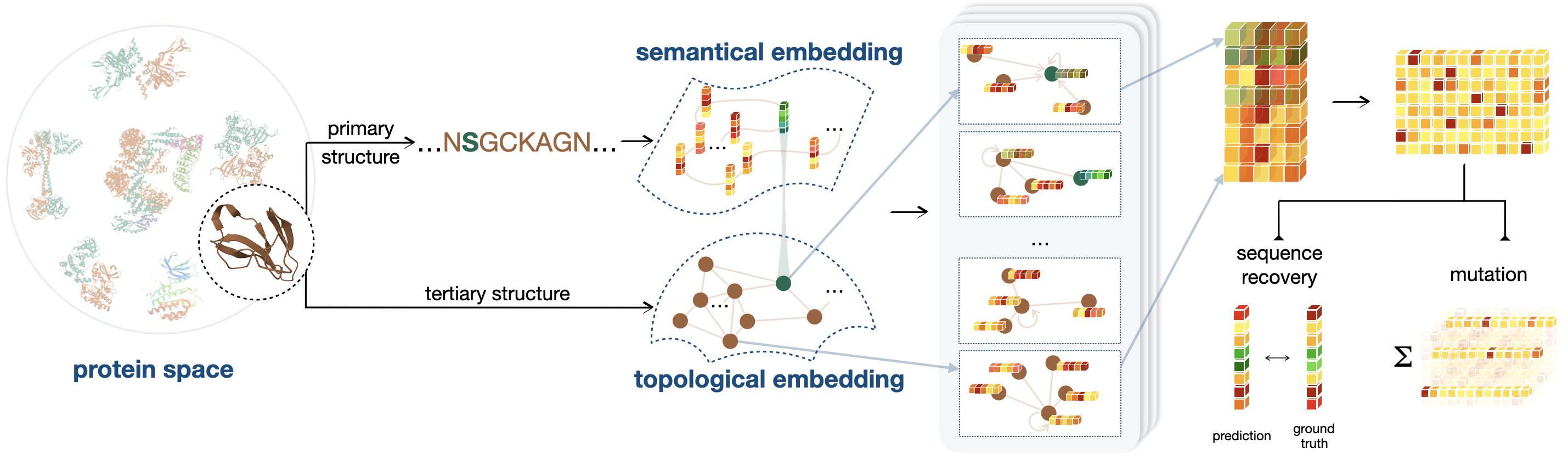
We focus on using end-to-end methods for protein directed evolution in zero sample scenarios, our overall framework can be shown in the following figure.
-
It introduces a novel pre-training framework that combines sequential and geometric encoders for protein primary and tertiary structures, guiding mutation directions by simulating natural selection on wild-type proteins.
-
Its framework demonstrates exceptional performance in predicting the effects of protein variants on bioactivity and thermostability by capturing the semantic and topological characteristics of proteins, significantly improving accuracy compared to existing methods.
-
It introduces two new benchmarks, DTm and DDG, specifically designed to assess protein thermostability, providing a detailed and practical evaluation environment that supplements existing datasets and enhances the understanding of model performance under various conditions.
-
ProteinGym Model:
The pdb files folded by ColabFold 1.5 can be downloaded here.
The pdb and mutant files in ProtSSN formation can be downloaded here.
We conducted tests on ProteinGym and compared it with the non-MSA method. Meanwhile, due to the fusion of structural information, we also conducted tests on two newly constructed stability datasets, DTm and DDG, in order to verify the correlation between structure and stability.

Our model surpasses most MSA methods and almost all language model methods without considering viruses, which means you can confidently use our model end-to-end. At the same time, because there is no need for MSA for reasoning or training, this reduces the tedious process and prior knowledge of collecting MSA, and there is no potential performance instability caused by MSA search methods/quantities.

DTM and DDG dataset can be found in data/DTM and data/DDG
Please make sure you have installed Anaconda3 or Miniconda3.
If you already have a enviroment.
We recommend that the torch_geometric should been updated to 2.3 or higher.
If you don't have a enviroment.
conda env create -f environment.yaml
conda activate protssn
pip install torch_scatter torch_sparse torch_cluster -f https://data.pyg.org/whl/torch-2.3.0+cu121.html- For direct use of inference, we recommend at least 10G of graphics memory, such as RTX 3080
- For retraining a ProtSSN, we recommend that the larger the graphics memory. We recommend a 24GB RTX 3090 or better.
We have prepared 9 models that you can use separately or integrate. 😉
If you only want to only use one model for reasoning, we recommend using k20_h512.
mkdir model
cd model
wget https://lianglab.sjtu.edu.cn/files/ProtSSN-2024/model/protssn_k20_h512.ptEnsembling models can achieve better results, and given our lower reasoning costs, you can use ensemble methods for zero-shot prediction. 😊
ProtSSN.tar contains all the model checkpoints, our training records and configs can be found in model/history and model/config.
wget https://lianglab.sjtu.edu.cn/files/ProtSSN-2024/ProtSSN.model.tar
tar -xvf ProtSSN.model.tar
rm ProtSSN.model.tarHere is a subset of ProtetinGym as the basic sample for mutation prediction in data/mutant_example, the files should be arranged in the following format:
data/proteingym-benchmark
|——DATASET
|——|——Protein1
|——|——|——Protein1.pdb
|——|——|——Protein1.tsv
|——|——Protein2
|——|——...
- Because our model requires structure (PDB) as input, we recommend using Alphafold for folding, and of course, ESMFold is also a good choice.
- If you do not have a tool installed for folding protein structures, you can search for your protein by Uniprot ID in the AlphaFold database (https://alphafold.ebi.ac.uk/) without consuming resources for folding.
The following script can be found at script/zeroshot_predict.sh, more details can be found at zeroshot_predict.py. We recommend using ensemble method for inference, which can be completed with a single RTX3080.
# k in (10, 20, 30)
# h in (512, 768, 1280)
# use single model for inference (default)
DATASET=proteingym-benchmark
CUDA_VISIBLE_DEVICES=0 python zeroshot_predict.py \
--gnn_model_name k10_h512 \
--mutant_dataset_dir data/mutant_example/$DATASET \
--result_dir result/$DATASET
# select the models for ensemble prediction
DATASET=proteingym-benchmark
CUDA_VISIBLE_DEVICES=0 python zeroshot_predict.py \
--gnn_model_name k10_h512 k20_h512 k30_h512 k10_h768 k20_h768 k30_h768 k10_h1280 k20_h1280 k30_h1280 \
--use_ensemble \
--mutant_dataset_dir data/mutant_example/$DATASET \
--result_dir result/$DATASETIn this case, we suggest that you start from a single point position, and you still need to organize your protein sequence and structural files in the same way as above.
You can use the following script to construct data for single point saturation mutations and use models for prediction.
python src/build_sav.py -d data/mutant_example/no_exp
# output: A0A5J4FAY1_MICAE contains 14193Perhaps you need protein embeddings for evolutionary or other direction analysis.
Here is a basic sample for embedding extraction in data/mutant_example, the files should be arranged in the following format:
data/proteingym-benchmark
|——DATASET
|——|——Protein1
|——|——|——Protein1.pdb
|——|——|——Protein1.tsv (no need)
|——|——Protein2
|——|——...
DATASET=proteingym-benchmark
CUDA_VISIBLE_DEVICES=0 python get_embedding.py \
--gnn_model_name k10_h512 k20_h512 \
--mutant_dataset_dir data/mutant_example/$DATASET \
--result_dir result/embed/$DATASETThe output file is <model>.pt which is the following format:
esm_embed is the embedding from facebook/esm2_650M, protssn_embed is the embedding from protssn which fusues the protein language and sturcture information.
{
protein1: {"esm_embed": esm_embed, "protssn_embed": protssn_embed},
protein2: {"esm_embed": esm_embed, "protssn_embed": protssn_embed},
...
}We use CATH for pre-training, you can find more details in https://www.cathdb.info/.
mkdir -p data/cath
cd data/cath
wget https://huggingface.co/datasets/tyang816/cath/blob/main/dompdb.tar
# or wget https://lianglab.sjtu.edu.cn/files/ProtSSN-2024/dompdb.tar
tar -xvf dompdb.tar
rm dompdb.tarYou can first create CATH graphs, but if you skip this step, the graphs will be automatically created in start training step.
see script/build_cath_dataset.sh
# k can be any number if you want
K=30
python src/data.py \
--data_type cath \
--c_alpha_max_neighbors $K \
--cath_dataset data/cath/cath_k$KYou can view our work as an adapter to enhance the capabilities of language models, so you can replace any language model as a prefix.
see script/run_pt.sh
K=20
H=512
CUDA_VISIBLE_DEVICES=0 \
python run_pt.py \
--seed 12345 \
--noise_ratio 0.05 \
--noise_type mut \
--gnn egnn \
--gnn_config src/config/egnn.yaml \
--gnn_hidden_dim $H \
--plm facebook/esm2_t33_650M_UR50D \
--cath_dataset data/cath/cath_k$K \
--c_alpha_max_neighbors $K \
--learning_rate 1e-4 \
--warmup_percent 0 \
--weight_decay 1e-4 \
--num_train_epochs 100 \
--batch_token_num 1024 \
--max_grad_norm 4 \
--gradient_accumulation_steps 10 \
--patience 20 \
--model_dir debug/model/"k"$K"_h"$HFor further questions, please feel free to contact Yang Tan.
This project is under the MIT license. See LICENSE for details.
If you find this repository useful, please consider citing this paper:
@article{tan2023protssn,
title={Semantical and Topological Protein Encoding Toward Enhanced Bioactivity and Thermostability},
author={Tan, Yang and Zhou, Bingxin and Zheng, Lirong and Fan, Guisheng and Hong, Liang},
journal={bioRxiv},
pages={2023--12},
year={2023},
publisher={Cold Spring Harbor Laboratory}
}