
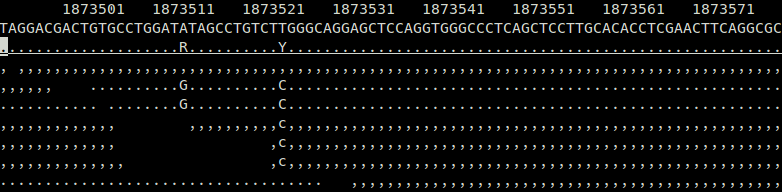
HHGA transforms genomic alignments and candidate haplotypes into a feature-space representation suitable for use by machine learning systems. We take inspiration from genome visualizers that are often used to inspect candidate variant calls. For example, samtools tview implements a text-based view of alignments in a particular region of the reference genome with encodings representing the read placement, direction, non-reference bases in the alignments, gaps (insertions and deletions are both shown faithfully using a gap character), and the qualities of mismatches. Note that the tview representation is matrix-like, but the default behavior of HHGA does not generate a true matrix.
Additional tools provide mechanisms to convert these representations into human-readable formats (both text and image) for debugging.
HHGA can be thought of as an example decision synthesizer. We use it in combination with genomic truth sets to generate examples from sequencing data that can be used in decision learning. Our primary interest is to approximate P(genotype|alignments), where the genotype is composed of N haplotypes (typically 2), and the alignments represent the observational data we have at the genomic locus.
We represent the MSA of the reads and the reference, the estimated per-base errors, the read strands releative to the reference, the alignment mapping qualities, and the candidate haplotypes in the genotype of the sample at the locus using the namespaced libSVM format used by Vowpal Wabbit.
As an example, if this is a tview-like representation of a set of alignments at a putative SNP site, augmented with candidate haplotypes:
ref: TAT
hap1: ...
hap2: .G.
aln1: ,,,
aln2: .G.
aln3: g,
aln4: ..We use six symbols to encode the MSA, { A, T, G, C, N, U, M }, where N is the degenerate symbol (it could represent any of A, T, G, or C), U is a gap symbol required to normalize the MSA into a matrix, and M is a symbol indicating if we don't have any information at the position in the MSA.
Mapping quality and the strandedness of the reads are represented in two namespaces (mapq and rev).
Assuming the genotype is true, there are a mixture of 3 different error probabilities for the bases in the reads, two of the reads are on each strand, and the mapping qualities for the alignments are aln1: 60, aln2: 20, aln3: 50, aln4: 30, the feature space transformation of this site would be:
1 |ref T:1 A:1 T:1
|hap1 T:1 A:1 T:1
|hap2 T:1 G:1 T:1
|mapq aln1:60 aln2:20 aln3:50 aln4:30
|strand aln1:1 aln2:1 aln3:1
|aln1 T:0.94 A:0.91 T:0.991
|aln2 T:0.94 G:0.991 T:0.94
|aln3 M:1 G:0.94 T:0.94
|aln4 T:0.94 G:0.94 M:1Newlines are included here only for legibility. This would be on one line of the output of HHGA.
As in libSVM format, the first entry in the output defines the class of the example. By convention, we say the example is 1 if the haplotypes are correct, and -1 otherwise.
Sketch of usage. We extract the feature space representation using windows of size 100 at sites that are defined in the candidates in the variant input file. We use truth.vcf.gz to indicate which genotypes and alleles are true. All others in vars.vcf.gz are assumed false.
hhga -t truth.vcf.gz -v vars.vcf.gz -w 100 -b aln.bam -f ref.fa | vw --save_resume --ngram 5 --skips 3 --loss_function logistic --interactions rhmsaThe output can now be used in Vowpal Wabbit and other systems which support the same input format.
Make this go, add a method to generate more training data per site by varying the length of the candidate haplotypes (e.g. full-window vs. just SNP view), add image output for debugging.