Here is the pipeline I used for ChIP-seq preprocessing, including:
- align the fastq data to reference genome by bowtie or bowtie2.
- run FastQC to check the sequencing quality.
- remove all reads duplications of the aligned data.
- generate TDF files for browsing in IGV.
- run PhantomPeak to check the quality of ChIP.
- run ngs.plot to investigate the enrichment of ChIP-seq data at TSS, TES, and genebody.
The pipeline work flow is:
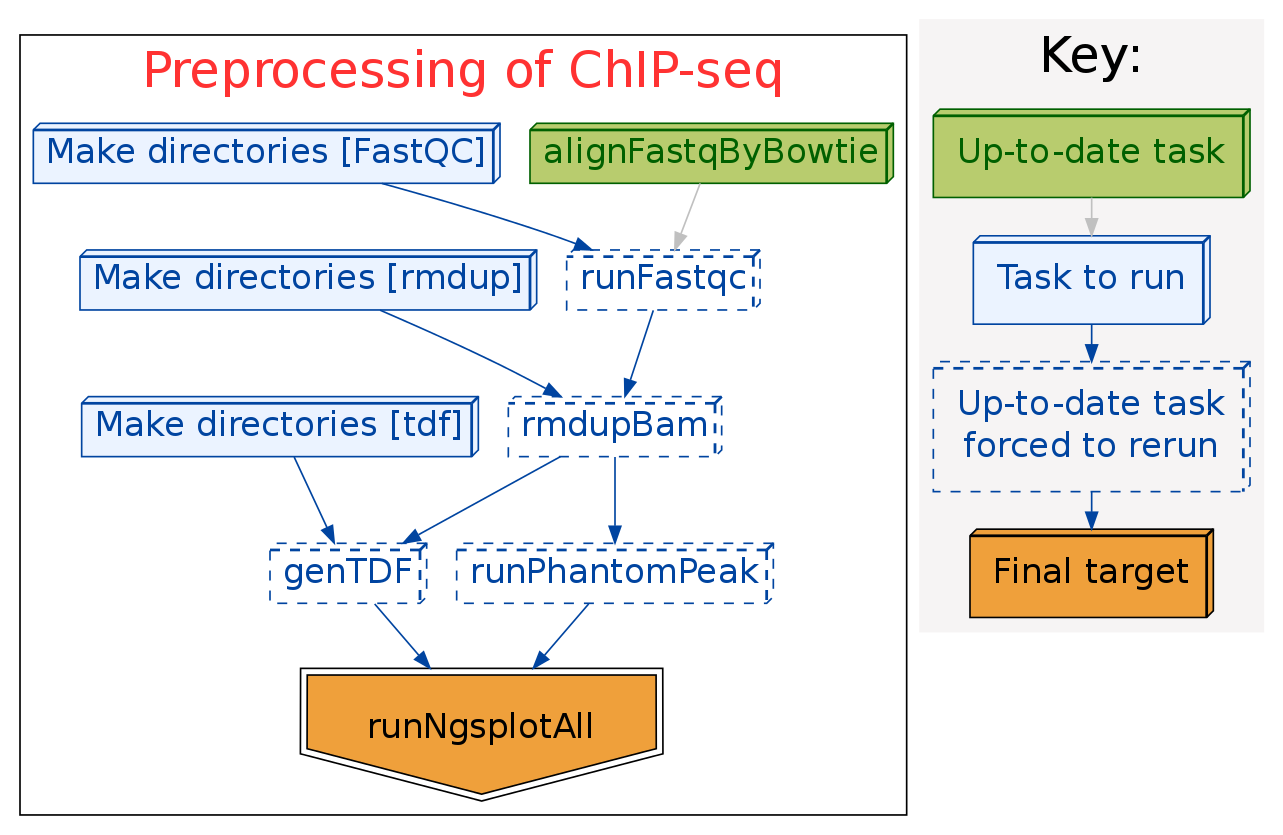
The softwares used in this pipeline are:
- ruffus
- Bowtie
- FastQC
- samtools
- IGVTools
- PhantomPeak In fact, the script run_spp_nodups.R is from PhantomPeak, but PhantomPeak still need to be installed in R.
- ngs.plot
- If cluster supporting needed, drmaa_for_python is needed. Now LSF and SGE are supported, but it is easy to modify it to fit your demands.
Install above softwares and make sure they are in $PATH.
Put the scripts in ./bin to a place in $PATH or add ./bin to $PATH.
python pipeline.py config.yamlOr on an LSF or SGE cluster:
nohup python pipeline.py config.yaml &If you want to stop at some step before runNgsplotAll, you may modify pipeline_run in pipeline.py, change the function name to the step you want.
After the running of the pipeline, then to summarize the result:
python results_parser.py config.yamlFor the organization of projects, I generally follow this paper: A Quick Guide to Organizing Computational Biology Projects. Here because it is preprocessing, and real analysis will be peak calling, chromatin segmentation, and differential enrichment detection, so I just put the results of the preprocess in the data folder.
For the configuration yaml file, project_dir: ~/projects/test_ChIP-seq and data_dir: "data" mean the data folder is ~/projects/test_ChIP-seq/data, and the results will be put in the same folder. Fastq files should be under ~/projects/test_ChIP-seq/data/fastq folder. Now *.fastq, *.fq, *.gz (compressed fastq) files are acceptable. aligner now could be bowtie or bowtie2, if not assigned, then default aligner is bowtie. For bowtie2, the system variable $BOWTIE2_INDEXES should be set before running.
The position of pipeline.py, results_parser.py, and config.yaml doesn't matter at all. But I prefer to put them under project/script/preprocess folder.
Important:
- To make ngs.plot part work, please name the fastq files in this way:
Say condition A, B, each with 2 replicates, and one DNA input per condition.
Name the files as A_rep1.fastq, A_rep2.fastq, A_input.fastq, B_rep1.fastq,
B_rep2.fastq, and B_input.fastq.The key point is to make the same condition
samples with common letters and input samples contain "input" or "Input"
strings.
- If use want to only run to some specific step, just modify the function name in
pipeline_runin pipeline.py. - If the data are pair-end, follow this step:
- Modify the
config.yamlfile, change "pair_end" to "yes". - Modify the
config.yamlfile, change "input_files" to "*R1*.fastq.gz" or "*R1*.fastq". - Make sure the fastq files named as "*R1*" and "*R2*" pattern.
- Modify the
- if you want to use cluster:
- Edit '~/.bash_profile' to make sure all paths in $PATH.
- Modify
config.yamlto fit your demands. multithreadinpipeline.pydetermines the number of concurrent jobs to be submitted to cluster nodes by ruffus. A default value of 10 is used.
Warning:
Bowtie2 allows multiple hits reads, and breaks the assumption of phantomPeak:
It is EXTREMELY important to filter out multi-mapping reads from the BAM/tagAlign
files. Large number of multimapping reads can severly affect the phantom peak
coefficient and peak calling results.
So be careful to interpret NSC and RSC in Bowtie2 alignment results.
The parameters of bowtie were just adopted from this paper.
In Bowtie2, default parameters are used.
- Method to skip some steps if the user doesn't run.