Mesoscopic Multiscale Simulations
Description
This Use Case for the OpenModel project describes the creation of a 2D-periodic membrane of poly-lactic-glycolic-acid (PLGA) surrounded by water (TIP4P 2005 model). The workflow starts with a coarse-grained force field based on the MOLC model, implemented in a fork of LAMMPS, version 30 Oct 2019. The sample is first assembled in the desired configuration, annealed, converted to atomistic coordinates with the program BACKMAP, and finally annealed with a type-I atomistic force field (OPLS). The resulting structure is suitable for ab-initio calculations (paper in preparation). However, this last step is already an established procedure and is not covered here.
The whole procedure is based on open-source software and relies on a coarse-grained force field parametrised consistently with the atomistic force field. Since LAMMPS has limited capabilities to handle complex structures, the strategy used here is to manipulate molecular samples with a custom-written program, dumptools, and to combine the structure with the force field using MOLTEMPLATE.
We acknowledge PRACE for awarding us access to the ARCHER2 UK National Supercomputing Service (https://www.archer2.ac.uk) hosted by at EPCC, UK.
Software Dependency
This Use Case depends on the following software. Additional scripts are included in the scripts folder.
- MOLTEMPLATE.
- LAMMPS 30 Oct 2019, including the USER-MOLC package.
- BACKMAP.
- VMD.
Workflow
Go to the multiscale folder and execute the following steps.
- Create a 2x2x1 supercell from the bulk model the polymer, turn it into a slab, and create the input files
for the first molecular dynamics simulation.
Amorphous bulk structure of PLGA 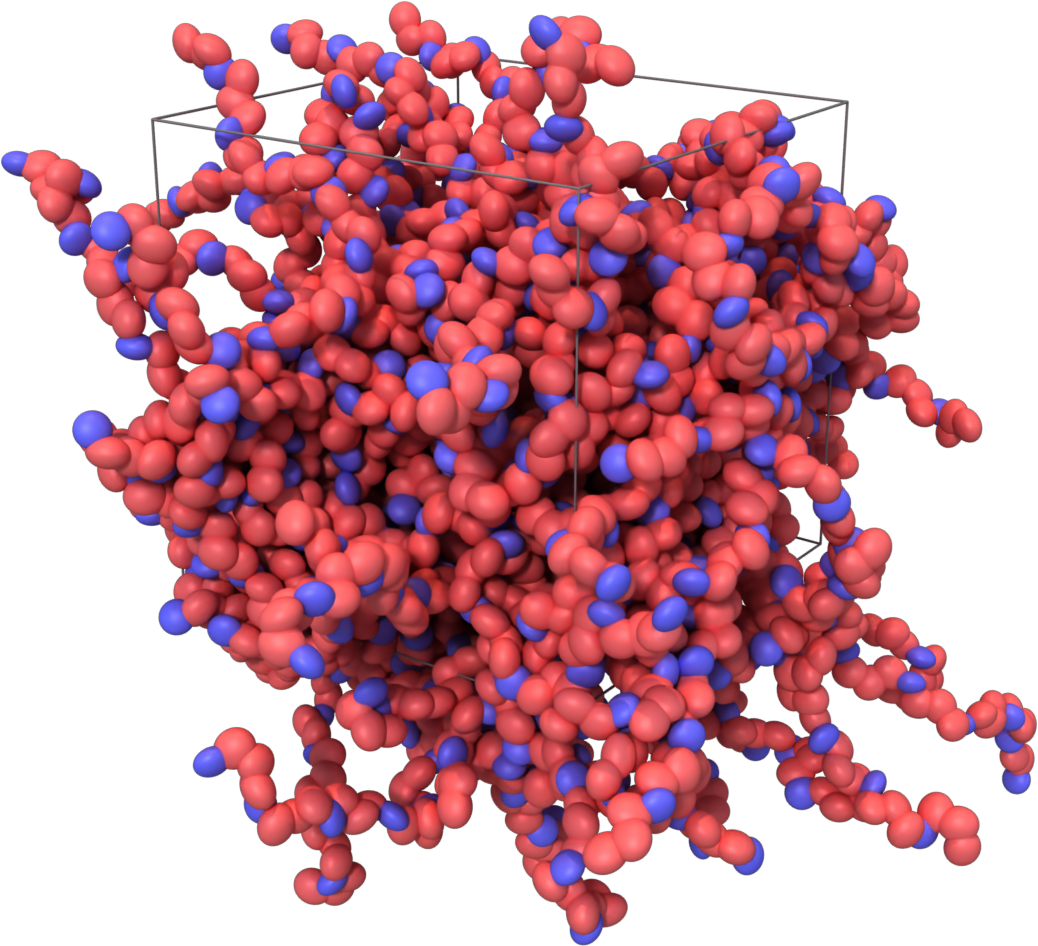
bash 01_preprocess.sh
- Anneal the slab in vacuum. LAMMPS is typically executed in parallel, depending on the architecture on which it has been compiled. A batch script for the SLURM workload manager is also provided as an example of usage on HPC facilities.
mpirun -np 128 lmp_bin -in 02_cg_slab_01.inInitial structure Relaxed structure 

- Create a supercell of solvent big enough to fill the simulation cell containing the slab. Trim the solvent supercell as to avoid bad contacts with the polymer slab. Write the input files for the next CG-MD simulation.
bash 03_postprocess.sh
- Relax the structure along Z (i.e. the direction perpendicular to the slab surface).
mpirun -np 128 lmp_bin -in 04_cg_sample_01.inInitial structure Relaxed structure 
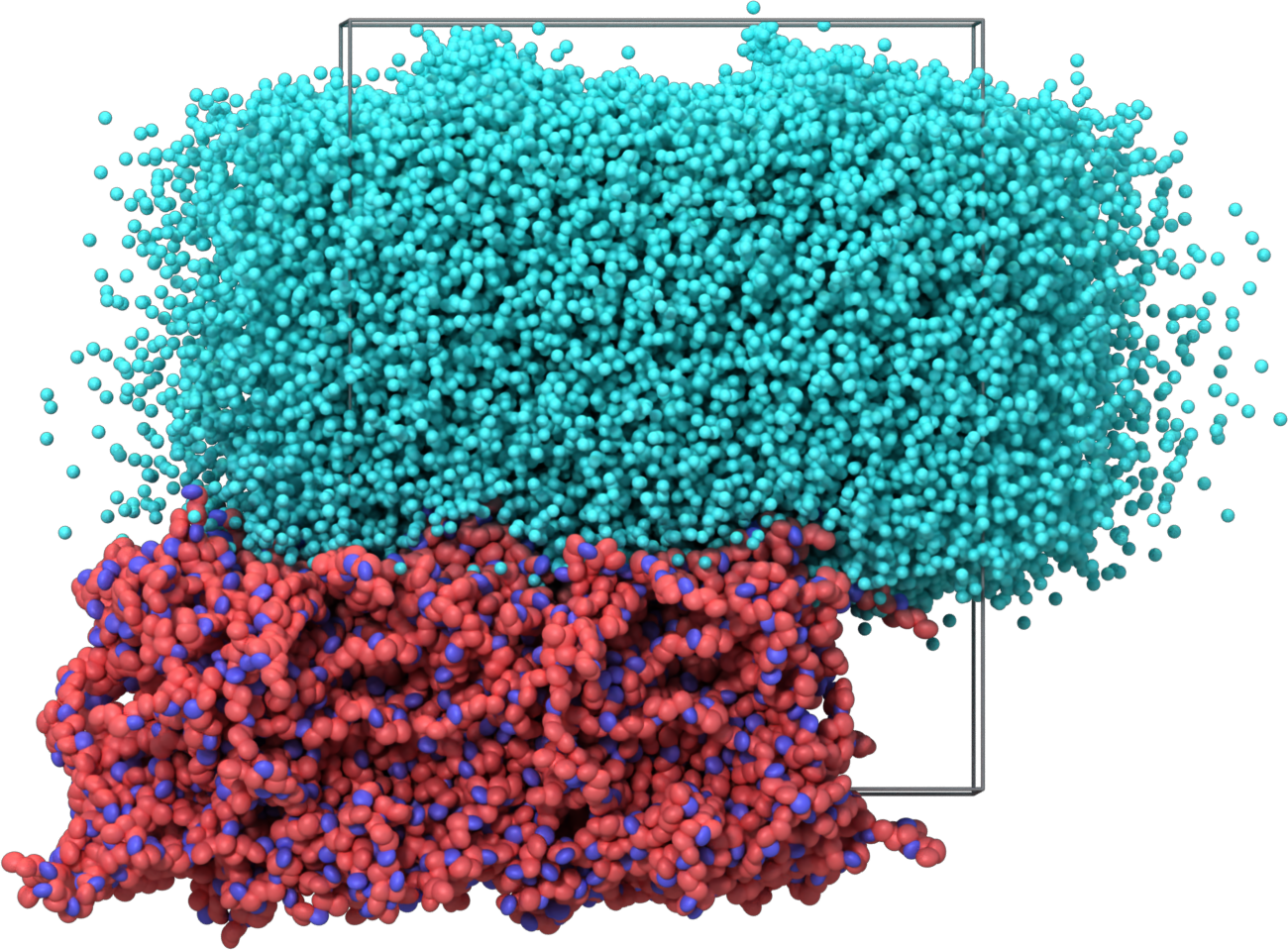
- Wrap the last frame of the CG trajectory and patch the bead types to distinguish the initial and final monomers in the polymer chain. Convert the wrapped structure to atomistic coordinates and write the input files for an all-atom MD simulation.
bash 05_postprocess.sh
Wrapped CG structure Backmapped atomistic structure 

- Anneal the structure at room temperature with a standard all-atom force field. This simulation is computationally quite expensive.
mpirun -np 1280 lmp_bin -in 04_cg_sample_01.in - Extract the last (relaxed) snapshot from the binary MD trajectory, and save it as a PDB file for later use.
bash 07_postprocess.sh









Notes
LAMMPS 30Oct19 has been compiled with the following packages enabled:
make yes-asphere yes-kspace yes-mc yes-misc yes-molecule yes-rigid yes-user-molfile yes-user-molc
make -j4 g++_openmpi