

| Test Status |
|
| Code Quality |


|
| Version Info |




|
| License |

|
| Documentation |
Compatibility Details
| Operating System | Python version |
|---|---|
| Linux | |
| MacOs | |
| Windows |
The histo-pathological analysis of tissue sections is the gold standard to assess the presence of many complex diseases, such as tumors, and understand their nature. In daily practice, pathologists usually perform microscopy examination of tissue slides considering a limited number of regions and the clinical evaluation relies on several factors such as nuclei morphology, cell distribution, and color (staining): this process is time consuming, could lead to information loss, and suffers from inter-observer variability.
The advent of digital pathology is changing the way pathologists work and collaborate, and has opened the way to a new era in computational pathology. In particular, histopathology is expected to be at the center of the AI revolution in medicine [1], prevision supported by the increasing success of deep learning applications to digital pathology.
Whole Slide Images (WSIs), namely the translation of tissue slides from glass to digital format, are a great source of information from both a medical and a computational point of view. WSIs can be coloured with different staining techniques (e.g. H&E or IHC), and are usually very large in size (up to several GB per slide). Because of WSIs typical pyramidal structure, images can be retrieved at different magnification factors, providing a further layer of information beyond color.
However, processing WSIs is far from being trivial. First of all, WSIs can be stored in different proprietary formats, according to the scanner used to digitalize the slides, and a standard protocol is still missing. WSIs can also present artifacts, such as shadows, mold, or annotations (pen marks) that are not useful. Moreover, giving their dimensions, it is not possible to process a WSI all at once, or, for example, to feed a neural network: it is necessary to crop smaller regions of tissues (tiles), which in turns require a tissue detection step.
The aim of this project is to provide a tool for WSI processing in a reproducible environment to support clinical and scientific research. histolab is designed to handle WSIs, automatically detect the tissue, and retrieve informative tiles, and it can thus be integrated in a deep learning pipeline.
Please see installation instructions.
Read the full documentation here https://histolab.readthedocs.io/en/latest/.
Join our user group on  Slack
Slack

Here we present a step-by-step tutorial on the use of histolab to
extract a tile dataset from example WSIs. The corresponding Jupyter
Notebook is available at https://github.com/histolab/histolab-box:
this repository contains a complete histolab environment that can be
used through Docker on all platforms.
Thus, the user can decide either to use histolab through
histolab-box or installing it in his/her python virtual environment
(using conda, pipenv, pyenv, virtualenv, etc...). In the latter case, as
the histolab package has been published on (PyPi),
it can be easily installed via the command:
pip install histolab
alternatively, it can be installed via conda:
conda install -c conda-forge histolab
First things first, let’s import some data to work with, for example the
prostate tissue slide and the ovarian tissue slide available in the
data module:
from histolab.data import prostate_tissue, ovarian_tissueNote: To use the data module, you need to install pooch, also
available on PyPI (https://pypi.org/project/pooch/). This step is
needless if we are using the Vagrant/Docker virtual environment.
The calling to a data function will automatically download the WSI
from the corresponding repository and save the slide in a cached
directory:
prostate_svs, prostate_path = prostate_tissue()
ovarian_svs, ovarian_path = ovarian_tissue()Notice that each data function outputs the corresponding slide, as an
OpenSlide object, and the path where the slide has been saved.
histolab maps a WSI file into a Slide object. Each usage of a WSI
requires a 1-o-1 association with a Slide object contained in the
slide module:
from histolab.slide import SlideTo initialize a Slide it is necessary to specify the WSI path, and the
processed_path where the tiles will be saved. In our
example, we want the processed_path of each slide to be a subfolder of
the current working directory:
import os
BASE_PATH = os.getcwd()
PROCESS_PATH_PROSTATE = os.path.join(BASE_PATH, 'prostate', 'processed')
PROCESS_PATH_OVARIAN = os.path.join(BASE_PATH, 'ovarian', 'processed')
prostate_slide = Slide(prostate_path, processed_path=PROCESS_PATH_PROSTATE)
ovarian_slide = Slide(ovarian_path, processed_path=PROCESS_PATH_OVARIAN)Note: If the slides were stored in the same folder, this can be done
directly on the whole dataset by using the SlideSet object of the
slide module.
With a Slide object we can easily retrieve information about the
slide, such as the slide name, the number of available levels, the
dimensions at native magnification or at a specified level:
print(f"Slide name: {prostate_slide.name}")
print(f"Levels: {prostate_slide.levels}")
print(f"Dimensions at level 0: {prostate_slide.dimensions}")
print(f"Dimensions at level 1: {prostate_slide.level_dimensions(level=1)}")
print(f"Dimensions at level 2: {prostate_slide.level_dimensions(level=2)}")Slide name: 6b725022-f1d5-4672-8c6c-de8140345210
Levels: [0, 1, 2]
Dimensions at level 0: (16000, 15316)
Dimensions at level 1: (4000, 3829)
Dimensions at level 2: (2000, 1914)
print(f"Slide name: {ovarian_slide.name}")
print(f"Levels: {ovarian_slide.levels}")
print(f"Dimensions at level 0: {ovarian_slide.dimensions}")
print(f"Dimensions at level 1: {ovarian_slide.level_dimensions(level=1)}")
print(f"Dimensions at level 2: {ovarian_slide.level_dimensions(level=2)}")Slide name: b777ec99-2811-4aa4-9568-13f68e380c86
Levels: [0, 1, 2]
Dimensions at level 0: (30001, 33987)
Dimensions at level 1: (7500, 8496)
Dimensions at level 2: (1875, 2124)
Note:
If the native magnification, i.e., the magnification factor used to scan the slide, is provided in the slide properties, it is also possible
to convert the desired level to its corresponding magnification factor with the level_magnification_factor property.
print(
"Native magnification factor:",
prostate_slide.level_magnification_factor()
)
print(
"Magnification factor corresponding to level 1:",
prostate_slide.level_magnification_factor(level=1),
) Native magnification factor: 20X
Magnification factor corresponding to level 1: 5.0X
Moreover, we can retrieve or show the slide thumbnail in a separate window:
prostate_slide.thumbnail
prostate_slide.show()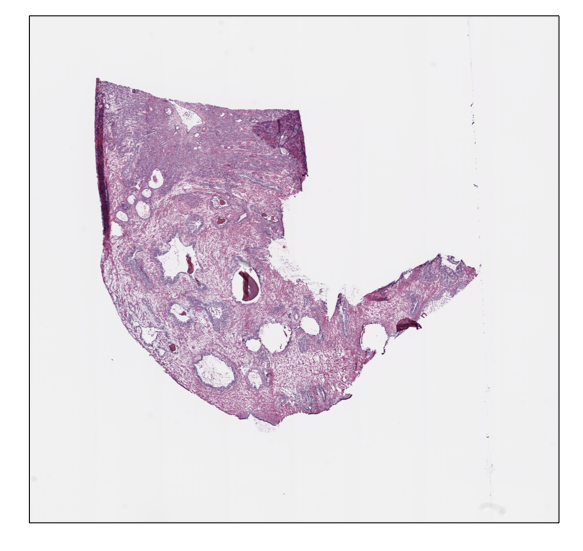
ovarian_slide.thumbnail
ovarian_slide.show()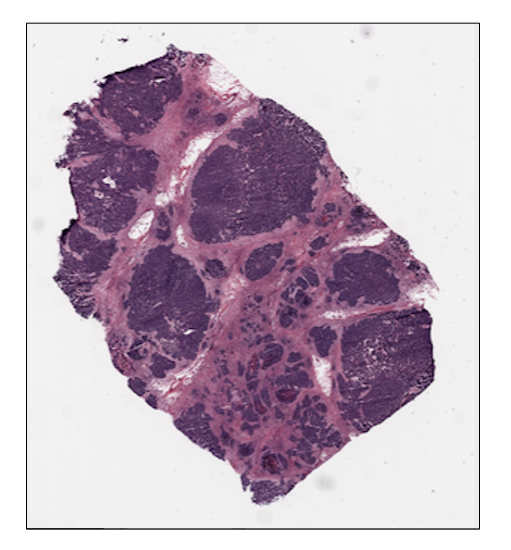
Once that the Slide objects are defined, we can proceed to extract the
tiles. To speed up the extraction process, histolab automatically
detects the tissue region with the largest connected area and crops the
tiles within this field. The tiler module implements different
strategies for the tiles extraction and provides an intuitive interface
to easily retrieve a tile dataset suitable for our task. In particular,
each extraction method is customizable with several common parameters:
tile_size: the tile size;level: the extraction level (from 0 to the number of available levels);check_tissue: if a minimum percentage of tissue is required to save the tiles;tissue_percent: number between 0.0 and 100.0 representing the minimum required percentage of tissue over the total area of the image (default is 80.0);prefix: a prefix to be added at the beginning of the tiles’ filename (default is the empty string);suffix: a suffix to be added to the end of the tiles’ filename (default is.png).
The simplest approach we may adopt is to randomly crop a fixed number of
tiles from our slides; in this case, we need the RandomTiler
extractor:
from histolab.tiler import RandomTilerLet us suppose that we want to randomly extract 30 squared tiles at level
2 of size 128 from our prostate slide, and that we want to save them only
if they have at least 80% of tissue inside. We then initialize our
RandomTiler extractor as follows:
random_tiles_extractor = RandomTiler(
tile_size=(128, 128),
n_tiles=30,
level=2,
seed=42,
check_tissue=True, # default
tissue_percent=80.0, # default
prefix="random/", # save tiles in the "random" subdirectory of slide's processed_path
suffix=".png" # default
)Notice that we also specify the random seed to ensure the reproducibility of the extraction process.
We may want to check which tiles have been selected by the tiler, before starting the extraction procedure and saving them;
the locate_tiles method of RandomTiler returns a scaled version of the slide with the corresponding tiles outlined. It is also possible to specify
the transparency of the background slide, and the color used for the border of each tile:
random_tiles_extractor.locate_tiles(
slide=prostate_slide,
scale_factor=24, # default
alpha=128, # default
outline="red", # default
)
Starting the extraction is then as simple as calling the extract method on the extractor, passing the
slide as parameter:
random_tiles_extractor.extract(prostate_slide)
Random tiles extracted from the prostate slide at level 2.
Instead of picking tiles at random, we may want to retrieve all the tiles available. The Grid Tiler extractor crops the tiles following a grid structure on the largest tissue region detected in the WSI:
from histolab.tiler import GridTilerIn our example, we want to extract squared tiles at level 0 of size 512
from our ovarian slide, independently of the amount of tissue detected.
By default, tiles will not overlap, namely the parameter defining the
number of overlapping pixels between two adjacent tiles,
pixel_overlap, is set to zero:
grid_tiles_extractor = GridTiler(
tile_size=(512, 512),
level=0,
check_tissue=False,
pixel_overlap=0, # default
prefix="grid/", # save tiles in the "grid" subdirectory of slide's processed_path
suffix=".png" # default
)Again, we can exploit the locate_tiles method to visualize the selected tiles on a scaled version of the slide:
grid_tiles_extractor.locate_tiles(
slide=ovarian_slide,
scale_factor=64,
alpha=64,
outline="#046C4C",
)
grid_tiles_extractor.extract(ovarian_slide)and the extraction process starts when the extract method is called on our extractor:

Examples of non-overlapping grid tiles extracted from the ovarian slide at level 0.
Depending on the task we will use our tile dataset for, the extracted
tiles may not be equally informative. The ScoreTiler allows us to save
only the "best" tiles, among all the ones extracted with a grid
structure, based on a specific scoring function. For example, let us
suppose that our goal is the detection of mitotic activity on our
ovarian slide. In this case, tiles with a higher presence of nuclei are
preferable over tiles with few or no nuclei. We can leverage the
NucleiScorer function of the scorer module to order the extracted
tiles based on the proportion of the tissue and of the hematoxylin
staining. In particular, the score is computed as where
is the percentage of nuclei and
the percentage of tissue in the tile t
First, we need the extractor and the scorer:
from histolab.tiler import ScoreTiler
from histolab.scorer import NucleiScorerAs the ScoreTiler extends the GridTiler extractor, we also set the
pixel_overlap as additional parameter. Moreover, we can specify the
number of the top tiles we want to save with the n_tile parameter:
scored_tiles_extractor = ScoreTiler(
scorer = NucleiScorer(),
tile_size=(512, 512),
n_tiles=100,
level=0,
check_tissue=True,
tissue_percent=80.0,
pixel_overlap=0, # default
prefix="scored/", # save tiles in the "scored" subdirectory of slide's processed_path
suffix=".png" # default
)Notice that also the ScoreTiler implements the locate_tiles method, which visualizes (on a scaled version of the slide) the first n_tiles with the highest scores:
grid_tiles_extractor.locate_tiles(slide=ovarian_slide)
Finally, when we extract our cropped images, we can also write a report of the saved tiles and their scores in a CSV file:
summary_filename = "summary_ovarian_tiles.csv"
SUMMARY_PATH = os.path.join(ovarian_slide.processed_path, summary_filename)
scored_tiles_extractor.extract(ovarian_slide, report_path=SUMMARY_PATH)
Representation of the score assigned to each extracted tile by the
NucleiScorer, based on the amount of nuclei detected.
We use PEP 440 for versioning.
This project is licensed under Apache License Version 2.0 - see the LICENSE.txt file for details
[1] Colling, Richard, et al. "Artificial intelligence in digital pathology: A roadmap to routine use in clinical practice." The Journal of pathology 249.2 (2019)
If you want to contribute to histolab, be sure to review the contribution guidelines