plastomics101
the handling editor Shu-Miaw Chaw
un bon papier redcube | une autre belle idee ici
comme quoi il faut juste une belle idee

## to hide the bootstrap values based on range
0,75 show=0
## to color bootstrap values based on range
80,90 color=red,size=12
100,120 color=darkred,size=14
Un papier interessant d'une equipe belge. I like the preprint image incorporating geographical and inflorescence type here is the final version. Mais l'espece endemique au Benin n'est pas dans la liste des especes utilisees Dracaena specksii
Echelle du temps

Donc l'apparition de Sesamum radiatum est vraiment recente.
Changer le titre
Cytogenetic and plastome phylogenomics indicate ascending dysploidy and recent hybridization as driven forces of speciation in Sesamum species complex
Abstract
- Enlever les parentheses avec les nombre de chromosomes au debut alors
- We also provided the first Fluorescence In Situ Hybridization (FISH) karyotyping of S. pedaloides revealing the chromosome number as 2n = 2x = 32. Comparative cytogenetic coupled with chroloplast genomes assessment unveiled that ascending dysploidy followed by hybridization contributed to the speciation in Sesamum genus.
Introduction
Among the studied species, the chromosome number of S. pedaloides was indertermined. Therefore, we took advantage of multifluor FISH karyotyping and whole.... for (i) comparative cytogenetic within Sesamum species (ii)...
Materials and Methods
Fluorescence In Situ Hybridization and Karyoptyping
To unveil the chromosome number set of S. pedaloides, a triple color FISH karyotyping was performed following [Nguyen et al method] (https://link.springer.com/content/pdf/10.1007/s13580-021-00364-9.pdf). Briefly, young and healthy root tips from S. pedaloides seedlings were collected and treated with 2mM of oxine at 12C for five hours. A Carnoy's solution was employed to fixed the pre-treated root tips at room temperature for two hours. The fixed root tips was then stored in 70% ethanol until the somatic chromosome spreads preparation step. Probes of Arabidopsis telomeric repeats, 5SrDNA, 45S rDNA were used for the FISH followed by the slide visualization with the Olympus BX53 microscope (Leica Microsystems Inc., Germany). The resulting image was processed with CytoVision v 7.2 (Leica Microsystem Inc., Germany) and Adobe Photoshop CS6. The same procedure was applied for Sesamum alatum (2n = 26) and Sesamum radiatum (2n = 64) for comparative cytogenetic analysis of the three set of chromosome number.
Results
FISH analysis
The FISH karyotype are presented in the Figure x.
The triple-color FISH karyotyping analysis resulted a total of 16 paired chromosomes for S. pedaloides. One pair of interstitial 5S rDNA locus (green) have been observed at chromosome pair No 5 and No 13 respectively. A set of three 45S rDNA loci (red) were detected at distal chromosomal regions at chromosome pair No 2, No 3, and No 4. While the majority of chromosome exhibited strong signals of Aradidopsis-type telomeric repeats (blue), weak signals were detected for the chromosome pair No 1, 10, 14, and 16.
S. alatum karyotype showed one strong pair of 45S rDNA loci at the chromosome pair No 10 while a colocalization of 5S rDNA and 45S rDNA was observed at the chomosome pair No 4.
As expected, S. radiatum karyoype revealed 32 pairs of chromosomes with clearly visible 45S rDNA signals (red) at the chromosome pairs No 1, 7, and 29. The chromosomes No 8, 9, and 13 exhibited 5S rDNA signals while a colocalization of 45S rDNA and 5S rDNA was observed at the chromosomes No 4and 5.
Discussion
Ascending dysploidy and hybridization events as speciation driving forces in Sesamum species complex
We provided for in the present study the first glance of S. pedaloides karyotype showing diploid genome pattern with 32 chromosomes, which is consistent with the tree topology. Meanwhile, the chromosome number variation from the diploid 2n = 26 to 2n = 32 is still a mystery. While the ancient species in the present study (S. alatum) and the cultivated type (S. indicum) are also diploid with 2 = 26 chromosomes, the group of 2n = 32 might have undergone chromosomal rearrangements, altering their karyotype. We speculate here that the 2n = 32 members including S. pedaloides, S. angolense, C. sesamoides (or S. sesamum), and C. triloba (S. trilobum) are the result of an ascending dysploidy event leading to their chromosome number change. Chromosome fission, fusion, or acquisition can leads to the increase of ploidy as termed as ascending dysploidy or, in contrast, descending dysploidy.Udall et al. 2019 Mayrose and Lysak 2021
As a consequence, this evolutionary event might be one of the source of speciation leading to the sister species C. sesamoides and C. triloba also known as S. sesamoides and S. triolobum. Similar ascending disploidy events have been also observed in Marantaceae Winterfeld et al. 2020 and Fabaceae Ta et al. 2021, Waminal et al. 2021 species complex.
Altogether, the present karyotyping provides a comprehensive preliminary data for cytotaxonomic studies in the Sesamum species complex.
Ajouter le volet hybridization apres.
Implications for diversity center of Sesamum
The present study exclusively dealt with African native Sesamum wild species. From our analyses, we speculate that since many wild sesame species are originated from Africa, the continent might be the origin of species diversification with the presence of diverse chromosome number sets. The cultivated species might occur later in the Indian sub-continent where the domestication took place (Figure x). Ethnobotanic studies are in agreement with this hypothesis since the 2n = 26, 2n = 32, and 2n = 64 wild species are widely used by local populations as medicine and food (Bedigian, 2018) while the cultivated type is predominantly employed in South East-Asian as food decoration (seeds) and therapeutic or nutraceutical (oil) complement (Miyake et al., 2005; Kim et al., 2016, 2020).
-
Perler dans la discussion de sesamum trilobum = Cerathoteca triloba et que Sesamum sesamoides = Cerathoteca sesamoides
-
Enlever novoplasty du document et agrder seulement getorganelle
Tables legends:
Table 1: Complete chloroplast genome statistics of wild sesame species.
Table 2:
Supplementary Table 1: Species collection sites
Supplementary Table 2: List of chloroplast genomes used in the present study
Supplementary Table 3: List of 75 chloroplast genomes genes used for phylogenetic tree inference
Supplementary Table 4: SSRs identification report in the assembled chloroplast genomes
Figures legends:
Figure 1: Map showing the geographical distibution of the studied species in their native (in green) and host habitat (in purple). (A): Sesamum indicum distribution. The native region covers Assam, Bangladesh, India, West Himalaya. It has a worldwide distribution. (B): Sesamum radiatum is native to Angola, Benin, Burkina, Cameroon, Cape Verde, Central African Repu, Chad, Congo, Gabon, Gambia, Ghana, Guinea, Gulf of Guinea Is., Ivory Coast, Liberia, Mali, Niger, Nigeria, Senegal, Sierra Leone, Togo, Zaïre. It has been introduced in Borneo, Guinea-Bissau, India, Malaya, Sri Lanka, Sumatera. (C): Sesamum pedaloides is originated from Angola and Namibia. (D): Sesamum angolense came from the Southern part of Africa covering Angola, Burundi, Kenya, Malawi, Mozambique, Namibia, Rwanda, Tanzania, Uganda, Zambia, Zaïre, and Zimbabwe. (E): Sesamum alatum was introduced in India. Its native distribution covers Angola, Benin, Botswana, Burkina, Cameroon, Caprivi Strip, Chad, Egypt, Eritrea, Ethiopia, Ghana, Guinea, Kenya, KwaZulu-Natal, Mali, Mauritania, Mozambique, Namibia, Niger, Nigeria, Senegal, Sudan, Swaziland, Western Sahara, Zambia, and Zimbabwe. (F): Cerathoteca triloba originated from Botswana, Cape Provinces, Free State, KwaZulu-Natal, Mozambique, Northern Provinces, Swaziland, and Zimbabwe. It has been introduced in Florida (US). (G): Cerathoteca sesamoides is native to Benin, Botswana, Burkina, Cameroon, Caprivi Strip, Central African Repu, Chad, Gambia, Ghana, Guinea, Guinea-Bissau, Ivory Coast, Kenya, Liberia, Malawi, Mali, Mauritania, Mozambique, Namibia, Niger, Nigeria, Senegal, Sierra Leone, Sudan, Tanzania, Togo, Uganda, Zambia, Zaïre, and Zimbabwe. Distribution data of each species were collected from POWO (2021).
POWO (2021). "Plants of the World Online. Facilitated by the Royal Botanic Gardens, Kew. Published on the Internet; http://www.plantsoftheworldonline.org/
Retrieved 15 December 2021."
Figure 2 vrai. FISH karyograms of the root metaphase chromosomes of Sesamum alatum (A), Sesamum pedaloides (B), and Sesamum radiatum (C). One pair of 5S rDNA and 45S rDNA colocalization (white arrow) were observed on the chromosome 4.
(A)-Two bands of 5S rDNA (green) signals were detected on the pericentromeric region of chromosome 4. Two pairs of intense 45S rDNA (red) signals was clearly seen in the shor arm of chromosome 4 (adjacent to 5S rDNA signal) and 10, whereas Aradidopsis-type telomere repeats signal was detected in the termini site of all chromosomes. (B)-Three pairs of 45S rDNA (red) were observed on the terminals regions of the chromosomes 2, 3, and 4, while two pairs of 5S rDNA (green) were detected on the short arm of the chromosomes 5 and 13. The absence of Arabidopsis-type telomere was noted on the chromosome 10 pair. (C)- Five pairs of 5S rDNA (green) signals were detected on the short arm of four homologous chromosomes, while one pair of signal was found in the paracentromeric region of the chromosome 13. Five pairs of intense 45S rDNA (red) signals hybridized on the terminals regions of the chromosomes 1, 4, 5, 7, and 29. Two pairs of 5S rDNA and 45S rDNA colocalization (white arrow) were observed on the short arm of the chromosome 4 and 5. Very week Arabidopsis-type telomere repeats signal was detected in the termini region of the chromosomes. All FISH karyograms were arranged from the longest to the shortest chromosomes, and paired according to FISH signals and size.
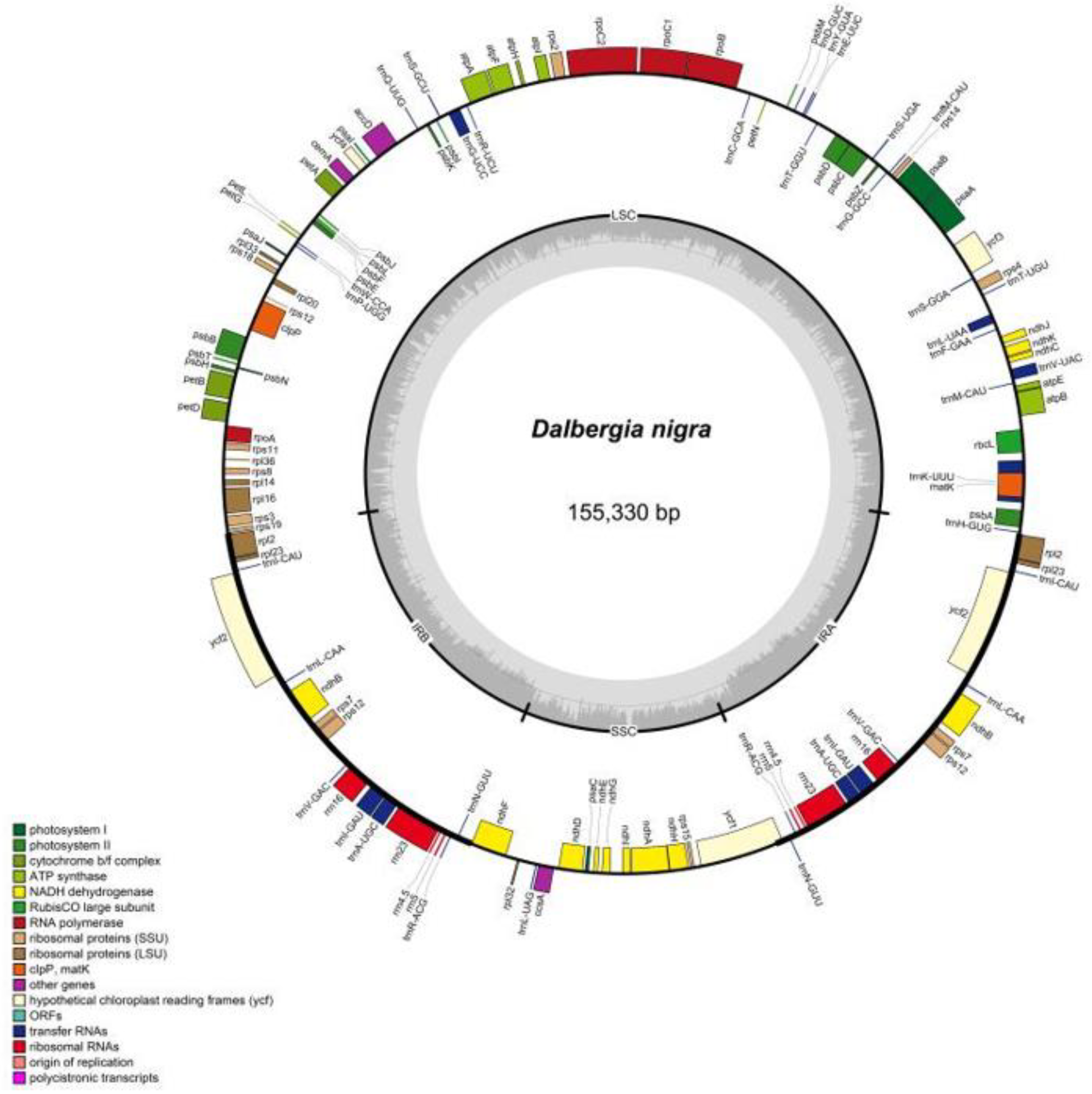
Figure 2: Chloroplast genome map of Sesamum and Cerathoteca species. Intron-containing genes are marked with asteriks symbol*. Dashed grey area in the inner circular map indicates the GC content. Different functionnal genes category are colored as indicated in the lower part of the genome map.
Figure 3: Phylogeny placement of newly constructed chloroplast genomes within Lamiales members. The phylogenetic tree was inferred from maximum likelihood and bayesian analyses of 75 conserved coding-genes. The closest Lamiales species cover Ancanthaceae and Linderniaceae species. Vitis vinifera was set as outgroup. Numbers above nodes are support values with maximum likelihood bootstrap values on the left, bayesian bootstrap values in the middle, and divergence times (in millions of years) estimated using RelTime approach implimented in MEGA X software. The newly assembled chloroplast genomes are indicated in blue.
Figure 4: Morphological and plastome-based comparative analysis between Sesamum and Cerathoteca species. (A): Phylogenetic tree inferred from maximum likelihood and bayesian analyses of 80 coding-genes. Bootstrap values were placed on each node with ML bootstrap values at left and bayesian bootstrap values at left. The ploidy, seed capsule anf flower photo are depicted in the center of the figure. (B): Alignment view of the chloroplast genomes using all-vs-all alignment approach implimented in AliTv software. The purple arrow represent the IR region. Each genome are colored following the link identity percentage as shown in right corner.
Figure 5: mVista view of the chloroplast alignment of seven Sesamum and two Cerathoteca species. The chloroplast genomes within the species are well conserved in both coding and non-coding regions.
Supplementary figure 1: Chloroplast genome alignment following progressivemauve alignment approach showing the conserved and divergent regions. The regions harboring the same color exhibit high similarities.
Figure 6: Chloroplast juctions sites view showing genes distribution alongside the boundaries and IR contraction and expansion within the Sesamum and Cerathoteca species. Arabidopsis thaliana was set as outgroup.
Figure 7: Nucleotide diversity variation with chloroplast genomes of Sesamum and Cerathoteca species. Highest values above 0.18 (in green) indicates candidates genes for population genetics purpose.
Figure 8: Evaluation of the discriminatory power of the candidate regions with the high nucleotide diversity index. The tree topology of ycf1 (A), psaC-ndhD (D), and ndhF (E) was consistent with the maximum likehood phylogenetic tree based on the 75 coding-genes. Discrepancies were noted for ndhA- and ndhE-based tree inference. Vitis vinifera was set as an outgroup. Squares and circles were colored following the ploidy and the bootstrap values respectively.
Figure 9: Heat map of relative synonymous codon usage (RSCU) values among Sesamum and Cerathoteca species.
Figure 10: Long-repeats count within Sesamum and Cerathoteca species chloroplast genomes.
Figure 11: Microsatellites count within Sesamum and Cerathoteca species chloroplast genomes
Figure 12: Microsatellite motifs count within Sesamum and Cerathoteca species chloroplast genomes
Figure 13: Heat map of pairwise Ka/ks values among wild relatives and the cultivated type Sesamum indicum. Non-applicable (with Ks = 0) Ka/Ks and ratio with Ks < 0.1 and Ks> 2 were changed into zero.
Code for evolview
## with legend;
!Title bootstrap
!Groups <=40,41~80,81~100
!Colors grey,gold,red
!LegendStyle circle
!bootstrapValueStyle show=1,style=circle
40 color=darkgrey
41,80 color=gold
81,100 color=red
Mots cles pour discuter Dated tribe-wide whole chloroplast genome phylogeny indicates recurrent hybridizations within Triticeae
Suivre simplement ceci pour rediger Comparative Analysis of Chloroplast Genomes of Seven Chaetoceros Species Revealed Variation Hotspots and Speciation Time
Results
Chloroplast genome general features
The assembled chloroplast genomes resulted in a single circular quadripartite genome with typical two IRs separeted by LSC and SSC (Figure 2. Chloroplast genome map). The plastome sizes were 153089 bp, 153096 bp, 153217 bp, 153285 bp, 153338 bp, 153287 bp, 153863 bp for S. angolense, S. alatum, S. pedaloides, S. radiatum, S. indicum cv Goenbaek, C. sesamoides, and C. triloba respectively (Table 1. Features of plastome genome). The seven genomes contained 114 unique genes including 80 coding-proteins, 30 tranfert RNA and four ribosomal RNA genes as previously found by Yi and Kim (2012) and Zhang et al. (2013) using S. indicum cv Ansanggae and S. indicum Yuzhi 11 as plant model respectively. Among the assembled chloroplast genomes, ten each coding sequence genes (atpF, rpoC1, rps12, petB, petD, rps16, rpl2, rpl16, ndhA, and ndhB) harbor a single intron while two coding genes contain two introns (clpP and ycf3). The GC contents were similar in IR (43.39 +/- 0.01 % ), LSC (36.40 +/- 0.03 %), SSC (32.62 +/- 0.08 %), and whole chloroplast genome scale (38.26 +/- 0.03 %) (Table 1. Features of plastome genome).
Phylogenetic Analysis of Sesamum and Ceratotheca Chloroplast Genomes
To figure out the evolutionary relationship between Sesamum, Cerathoteca, and the closest related species belonging to Acanthacae and Linderniaceae, a tree (Figure tree) was constructed using 75 shared coding protein sequences. Vitis vinifera was employed as an outgroup. As suspected, Sesamum and Cerathoteca representatives clustered in a single clade, resolving the sister species relationship. A close view of this Pedaliaceae clade highlihthed four major sub-clades. The sub-clade 1 represents a mixture of two ploidy levels with S. radiatum (2n = 64), S. angolense (2n = 32) and C. sesamoides (2n = 32). Interestingly, the sub-clade 2 encompassed S. pedaloides (2n = unknown) and C. triloba (2n = 32), suggesting S. pedaloides might also have 2n = 32 chromosomes. The sub-clade 3 grouped the cultivated species S. indicum (2n = 26) while the sub-clade 4 clearly spotted the wild relative S. alatum (2n = 26) as genetically distant from the cultivar, and other wild relatives. For additionnal investigation, we constructed a tree based on 80 coding sequence genes shared by our taxon of interest (Figure tree seven). As a result, the three topology was consistent with the previous one confirming the intricated relationship among both genus with.
Time divergent analysis
In order to understand the speciation occurrence time among Sesamum and Cerathoteca, a time divergence analysis was performed (Fiugure tree with time divergence). Firstly, S. alatum (Sub-clade4 in the tree), the unique seed with winges in our plant material, have occurred 14.46 Million years ago (Mya). Secondly, the sub-clade 2 members (Sesamum pedaloides and C. triloba) split concomitantly from the Sub-clade 1 and 2 block at about 5.26 Mya. Thirdly, C. sesamoides (member of the sub-clade1) was inferred to have occured 6.11 Mya, a little bit early than S. pedaloides and C. triloba. As expected, the cultivated type (S. indicum) have been occured lastly at 0.01 Mya. Interestingly, the time divergence inference revealed that S. radiatum (2n = 64) and C. sesamoides (2n = 32) have concomitantly occured recently at about 0.05 Mya. Taking together, the time divergence inference indicates recent hybridization between C. sesamoides (2n = 32) and S. angolense (2n = 32) leading to the new species S. radiatum (2n = 64).
Comparative plastome analysis
Do like here | and here and here
Although the morphological features of the species are distinct, the chloroplast genome structure is hihgly conserved (Figure AliTv). When comparing the plastid genome structure using mVista (Shuffle-LAGAN) (Figure x), MAuve (progressiveMauve alignment) (Figure y) and AliTv (lastz all-vs-all alignments) (Figure z), the conservative genome structure in coding and non-coding regions was also confirmed. However, IR contraction and expansion has been revealed with length ranging from 25096 bp to 25190 bp while LSC varied from 84872 bp to 85185 bp. SSC sized from 17719 bp to 17874 bp.
By comparing the chloroplast genomes boundaries of Sesamum and Cerathoteca species, we noted that the IRb/LSC junction is sited between rpl2 and rsp19 genes (Fgure IRscope). The pseudogene ycf1 is located exclusively in IRb for S. indicum cv Ansanggae, S. indicum cv Yuzhii11 and at the border IRb/SSC the the other species. The ndhF genes is maily in the SSC region for all species exepted for S. angolense. The ycf1 genes of all species were located at the SSC/IRa junctions with a gene size ranging from 5312 to 5369 bp. The trnH genes were localized in the LSC region, 1-16 bp away from the the IRa-LSC border. Overall the chloroplast genome structure at different junctions was highly conserved among Sesamum and Cerathoteca species.
Variation Hotspots within Chloroplast Genomes of Sesamum and Cerathoteca species
Despite the high-colinearity of the chloroplast genomes within Sesamum and Cerathoteca species, substancial variations were noted mainly in SSC regions (Figure Nucleotide diversity). The nucleotide diversity calculation revealed a peak value located in the ycf1 followed by ndhA, ndhE, psaC-ndh-D and ndhF regions. To estimate their discrimiatory power, we inferred the phylogenetic tree using each gene. As a result, ycf1, psaC-ndhD, and ndhF genes clearly distinguished the taxon as depicted earlier with a set of 75 and 80 coding genes respectively. Therefore, they could be used as a marker to delineate Sesamum and Cerathotheca species since several species are not yet well characterized at both morphologic and cytogenetic levels. Besides, C. sesamoides and S. radiatum exhibited a consistent monophylentic pattern for all divergent genes except ndhE, confirming the evolutionnary relationship (hybridization) between these two specific species.
Codon usage analysis
Codon usage bias was examined by computing the Relative Synonymous Codon Usage (RSCU) (Sharp and Cowe (1991)). RSCU represents the observed frequency of a codon divided by the expected frequency. A lack of bias referred to those codons with RSCU values close to 1. Globally, slight variation of RSCU were found within Sesamum and Cerathoteca species (Figure Heat map of relative synonymous codon usage (RSCU) values among Sesamum and Cerathoteca species.). A total of 27 core codons exhibited RSCU > 1, of which 24 were adenine/thymine-ending codons, one guanine-ending codons and two cytosine-ending codons. In contrast, guanine- or cytosine-ending codons mostly exhibited RSCU < 1. The most biased codon was found for the stop codon TAA (RSCU = 1.55 +/- 0.01) while the less biased was detected for the stop codon TAG (RSCU = 0.69 +/- 0.01). Similar trend of A-T biased condon usgae was observed for others plant species Biju et al 2019, Wang et al., 2017.
Interestingly, by examining the heatmap with species cluster, the phylogenetic tree topology is concordant with the coding sequence based tree inferencwe; indicating the robust estimation of phylogenetic relationship based on codon usage as observed in a wide range of families Wu et al. 2021 (Chi et al 2020), (Gao et al 2019), (Wang et al 2016)
Long and Simple Sequence Repeats
Long repeats constitute a driving force for chloroplast genome rearragement and has been used for phylogenetic inference between species Park et al. 2017-Luo et al. 2021. It induces genetic diversity by promoting intermolecular recombination in the chloroplast genome Park et al. 2018. Long repeats encompass forward,reverse, palindrome, and complement types. In the present study, the mean count of long repeats was 25.71 +/- 3.24 bp. The number of long repeats ranged from 21 to 31, among which palindromic (12-18) and forward (9-13) types are the most abundant. Besides, the size of the repeats was mainly within the range of 30–39 bp. Only S. indicum exhibited repeats in the range 60-69 bp (Figure long repeats).
Microsatellites are referred to short tandem repeat sequences of one to six nucleotidsrepeats (Fan et al 2007). SSRs are widely present in chloroplast genome and have been extensively used as molecular markers for population genetics, phylogenetic relationships inference, and species identification Powell et al 1995 Li et al 2020, Huang et al. 2018, Lee et al, 2018. We counted 21 to 32 chloroplast SSR within the assembled chloroplast genomes (Figure telle A), of which most are monomeric(> 87%) (Figure telle B). The majority (13-25) of SSRs are located in LSC sequence (Figure telle C) and mainly in intergenic regions (>76%) for all species (Figure telle D). The most dominant motif (A) count ranged from 19 to 27 and spaning 208-277 bp (Figure motif). Trinucleotide repeats (AAT) were only detected for S. alatum occuping 30 bp of the chloroplast genome length (Figure motif).
Selective pressure analysis
The pairwise ratios of the rate of non-synonymous substitutions (Ka) to the rate of synonymous substitutions (Ks) analysis is presented in the Figure xxx. Ka/Ks ratios with Ks < 0.1 or K > 0.2 were changed into zero for a reliable estimation of the selection pressure. The results revealed that the NAD(P)H-quinone oxidoreductase subunit I (ndhI) has undergone strong positive selection in S. angolense and S. radiatum. Similar trends were observed for the photosystem I gene ycf4 mainly in S. radiatum, S. alatum and C. sesamoides. The rpl20 gene also exhibited positive selection only in S. alatum. However, the matK, ndhF, and ycf1 Ka/ks values were around 1, implying neutral selection pressure.
Discusion suite one word on time divergence
In this study, Cerathoteca is robustly resolved as sister species species of Sesamum genus. Based on the time divergence infrence, we observed that S. radiatum and S. angolense appeared almost concomitantly alongside 2 = 32 representatives (C. sesamoides more specifically); suggesting the hydbridization event leading to 2n = 64 might involved both Sesamum angolense and Cerathoteca sesamoides. To test this hypothesis, we aligned long-reads high contiguous genome of S. angolense, ....
Inclure ceci avant le fait des alignment
Interestingly, a relatively long branch of S. alatum was observed implying its long evolutionary occurence compared to the other ones; which is consistent with tree topology from Gormley et al. 2015, biogeographic and morphological data [Ref Bedigian], and our time divergence estimation inference (14.46 MYA).
Globaly from the present study, we speculate that since many wild sesame are originated from Africa, the continent might be the origin of species diversification with the presence of 2n = 26, 2n = 32 and 2n = 64 species. The cultivated species might occured later on the Idian sub-continent where the domestication took place (Figure). Ethnobabic studies revealed that the 2n = 26, 2n = 32, and 2n = 64 wild species are widely used by local populations as medicine and food (Bedigian, 2019) while the cultivated type is predominently employed in South East-Asian as food decoration (seeds) and therapeutic or nutraceutical (oil) complement (Kim et al. 2016; Kim et al 2020, Miyake et al 2005). Although S. malabirum, an Indian native wild is suggested as progenitor Pathack et al. 2015, genomics-based evidence is required since many others Sesamum species are still not well characterized.
Revoir la section family and tribe les trois familles donnees dans le texte sont des tribes attention attention corriger dans le texte
Discussion
Plastome evolution between Sesamum and Cerathoteca genus
We reported the first whole chloroplast genome of six African native wild sesame species including S. alatum, S. angolense, S. pedaloides, S. radiatum, C. sesamoides, and C. triloba. The information from the generated plastome sequences served for comparative analysis. A typical quadripartite chloroplast structure including LSC, SSC and two IRs was observed. In-depth comparative analysis revealed contraction and expansion events within all species. IR expansion and contraction of chloroplast parts is a common phenomenon observed in land plants resulting in the variation of chloroplast length at both intra- and inter- species level (Ref). As expected, the chloroplast genome was highly converved among Sesamum and Cerathotheca despite the difference of systematic nomenclature. The conserved structure is consistent with the previously published S. indicum chloroplast genomes (Ref).
The variation of microsatellites copy numbers in chloroplast genome is helpful for population genetics, and polymorphysm assessment. Single-nucleotide repeats SSR datasets provided in the current study constitute a usefull resource for further population polymorphism study within Sesamum, Cerathoteca and potentially close relative species in Pedaliaceae family. Moreover, we detected that palindromic repeats are prominent in all chloroplast genomes. Palindromic repeats are known to constitute mutational hotspots contributing to plastome expansion [Can green algal plastid genome size be explained by DNA repair mechanisms? Genome Biol. Evol. 12, 3797–3802. doi: 10.1093/gbe/evaa012]. Therefore, they represent a suitable resource for genetic diversity investigation in Sesamum species continuum.
Chloroplast genes generally evolved under purifying selection mainly to maintain functionnal continuity of the genes over long period of time (Ref 1: Whole chloroplast genome comparison of rice, maize, and wheat: Implications for chloroplast gene diversification and phylogeny of cereals, Ref 2: Positive Selection Driving Cytoplasmic Genome Evolution of the Medicinally Important Ginseng Plant Genus Panax). However, previous plastome genomics studies identified some genes that underwent positive selection including photosynthetic genes rbcL and ycf2 for example (Ref rbcL Chloroplast Genome Analysis of Resurrection Tertiary Relict Haberlea rhodopensis Highlights Genes Important for Desiccation Stress Response, Ref ycf2 The complete chloroplast genome of the Jerusalem artichoke (Helianthus tuberosus L.) and an adaptive evolutionary analysis of the ycf2 gene).
In our study, mainly photosynthesis related genes including ndhA, ndhl, and ycf4 exhibited consistent positive selection. Subsequently, owing the specific distribution patterns of the studies samples in tropical Africa (See Figure 1) and the drought-prone habitat preference in nature, we postulate that the selection of these category of genes might related to adaptation to environment changes including photosynthetic rate, drought, temperature, carbone dioxyde level, or ecological niche. Moreover, the capability of photosynthetic-oriented genes selection may contribute the drought tolerance strenght of the cultivated sesame S. indicum as revealed by previous extensive functionnal genomics studies (Ref1 The genetic basis of drought tolerance in the high oil crop Sesamum indicum, Ref2 Insight into the evolution and functional characteristics of the pan-genome assembly from sesame landraces and modern cultivars)
Not sure but if ref needed use this [one](One-third of the plastid genes evolved under positive selection in PACMAD grasses)
Change S. pedaloides chromosome number is unknown
grosse decouverte sur sesamum angolense genre pedalium Aptera apparente au Cerathoteca ...blablabla
Includethis in comparative plastome section
Arabidopsis thaliana chloroplast genome (Genebank accession: NC_000932.1) was included as outgroup.
Include this in the phylogenetic tree section
Cholorplast genome datasets from close relative species belonging to Acanthacae and Linderniacae (Table x) were retrieved from NCBI. Vitis vinifera was used as outgroup. A total of 75 common coding protein genes (Supplementary table x: gene_per_species excel file) served for the phylogeny and time divergence inference.
Discussion
Our data provided a high-resolution view for delineation of pedaliaceae species in the sesame speciation continuum
Delineating species is quite challenging aspect in taxonomy since the methodolgy is quite heterogenous depending on not only of the taxa but also the scientists. Pedaliaceae family exhibits a complex species encompassing Sesamum, Josephinia and Cerathoteca species. Earlier tentative delineation using trnLF, ndhF, and ETS datasets revealed that Sesamum tribe (in Pedaliaceae family) is not monophyletic in contrast with other Lamiales representatives belonging to Martyniaceae and Plantaginaceae families. In fact, the authors highlithed that Pedaliaceae s.l. contains three families including Sesamothamneae, Sesameae, and pedalieae.
IR contraction DNA barcoding with example of the well known application good topology with respect to old data sets ka ks however ndh gene is stable but divergent gene
rna editing sites phenotyic plasticity, genetic diversity, speciation force driver of phenotypic plasticucity
Interspecific hybridization might drives speciation in Sesamum genus through chromosome number change
hybridization event is widely frequent in Planta lresulting in the occurence of multiple species() . In regards with the number of chromosome set from ourin the present study, a clear classification rised with grouping diploid 2n = 26 as well as putative polyploid 2n = 32 and 2n = 64. More importantly, S. radiatum, S. angolense and C. sesamoides form a clan suggesting hybridization event resulting into the advent of the S. radiatum species (2n = 64). This observation is in line with Karyotyping and on field hybridization test previously perforemed. Moreover, whole genome alignment of near chromosomal-grade assemblies of these five wild species (unpublished data) confirmed the presence of two sub-genomes from S. radiatum with the sub-genome A as Cerathoteca sesamoides and the subgenomeB as S. angolense. a west African cou try ain the same area. Therefore, the we postulate that the nomenclature C. sesamoides might change into Sesamum sesamoides. However, this conclusion is temporary since In thin sesamum family has not been clearly investigated for their chromosome set.e absence of others sesamum representative
Limits and prospects
The present study has the merit to provide dataset for elucidating the delineation between Sesamum and Cerathoteca. However,owing the large diversity observed in Sesamum, additionnal genomic datasets from Indian subcontinent including potential progenitor and others relatives species in Pepedaliaceae will help to resolve the origin of Sesamum indicum as well as a clear delination among Pedaliaceae s.l. members. Moreover, inaccurate species boundaries can negatively impact fundamental and applied research. Therefore speciation genomics at large scale in Pedaliacae family might shed light on speciation patterns in this singular group. Our study pave the way for eluctowards the elucidation of pedaliaceae family.
Interspecific hybridization mecanism of plant speciation homoploid hybrids allopolyploid
popart: full-feature software for haplotype network construction
May be used for but the number here is very small however may be take ploidy-wise
Homoploid hybrid species, which have no change in chromosome number relative to their parents, offer an opportunity to study the genomic consequences of hybridization in the absence of change in ploidy.
Hybridization can result in allopolyploid individuals, in which hybridization occurs at the same time as chromosome doubling, as well as homoploids, in which there is no change in chromosome number
affaire de copy number
Complete Chloroplast Genome Sequences and Comparative Analysis of Chenopodium quinoa and C. album
Comparative Analyses of Five Complete Chloroplast Genomes from the Genus Pterocarpus (Fabacaeae)
J'avais find un papier avant fait par la compagnie Phyzen. Le voici. Je pourrai publier le mien aussi dans Frontiers in Plant Science Section Evolutionnary and population Genetics IF 5.753 en 2021
Un autre avec une autre approache. Tres interessant pour argumenter plus tard.
Highly Diverse Shrub Willows (Salix L.) Share Highly Similar Plastomes
sesamum species list | Here is the folower of Sesamum triphyllum

BarcodingR: an integrated r package for species identification using DNA barcodes
4.3. DNA Barcode Development DNA barcodes have been widely used in evolutionary and phylogenetical studies for both plants and animals [49]. We compared cp genomes of Castanea using slideAnalyses to calculate genetic distance across a moving 800 bp window [47]. This method identified five regions of the Castanea genome that might be useful for barcoding: trnK-UUU-rps16, psbM-trnD-GUU, rbcL-accD, petA-psbJ, and rpl2 (Figures 4 and 5). We suggest the sequence of rbcL, matK, petA-psbJ as a barcode. The lengths of the three selected sequences are respectively 1428 bp, 1508 bp, and 1072 bp. There are 4, 14, and 75 mutation sites and 4, 10, and 68 informative sites within these loci. The barcode would be useful for distinguishing American chestnut (C. dentata) and Chinese chestnuts, and as such could provide a method to track the source of hybrids used in breeding. Simple sequence repeats (SSRs) were highly variable among and within Castanea species. As such, these have value for population genetics and other applications requiring high levels of polymorphism
Comparative Analysis of the Complete Chloroplast Genomes of Four Chestnut Species (Castanea)
MCMCTree inference
After doing multiple sequence alignment , trimming and potting the tree with MAFFT , tRIMaL AND iq-tREE . i HAVE CHECK WITH fIGTREE THE POSITIONNING AND THEN SET THE CLOCK INTERVAL. tHANKS TO TIMETREE.ORG FOR PROVIDING THIS WEBSITE. aFTER CHECKING i MANUUALLY SET SIT IN THE TREE LIKE THIS
16 1
((((tco,tbe),tfo)'B(0.41,0.66)',((((((sra,ago),cse),(spe,ctr)),((syu,sgo),san)),sal)'B(0.34,0.70)'(eat,(elo:,eon)))),vit);
Key note. first line should define the num of taxon ...here 6 and 1 for number of tree I think... Not sure...
the second file that we need is the sequence alignment file in some like file.....I use this website to convert in nexus file http://phylogeny.lirmm.fr/phylo_cgi/data_converter.cgiike
then I mannually set thet he format by removing the header and just get this
16 55609
vit atggaaaaatggtggttcaattcgatgttgtctaacgagaagttagaatacaggtgtgggctaagtaaatcaacggacagtcctgatcctaccagtggaagtgaagaccgggttataaataatacggataaaaacattaataattggagtcaaattaagagttctagttatagtaatgttgatcatttattcggcatcagggacattcggaatttcatctctgatgacacttttttagttagggataggaatggggtcagttatttcatatattttgatattgaaaatcaaatttttgagattgataacgatcagtcttttttgagtgaactagaaagttttttttatagttatcgaaattctagttatctgaataatgtatctaagagtaatcatccccactatgatcgttacatgtatgatactaaatatagttggaataagcacattaatagttgcattaataattacattt---------------------
The key point here is that the first line should provide the information relative to the number of taxon and the number of nucleotide
then in the second line, the name of the taxon 2 or more space the the sequence itself...
do the same for the remaining sequences....
then set the mcmc.ctl file
I chage a little bit by just change this .......cleandata = 0 to this .......cleandata = 1
And I got thos
seed = -1
seqfile = test1/sequence.txt
treefile = test1/tree.txt
outfile = out1
ndata = 1
seqtype = 0 * 0: nucleotides; 1:codons; 2:AAs
usedata = 1 * 0: no data; 1:seq like; 2:use in.BV; 3: out.BV
clock = 3 * 1: global clock; 2: independent rates; 3: correlated rates
RootAge = <1.0 * safe constraint on root age, used if no fossil for root.
model = 0 * 0:JC69, 1:K80, 2:F81, 3:F84, 4:HKY85
alpha = 0 * alpha for gamma rates at sites
ncatG = 5 * No. categories in discrete gamma
cleandata = 1 * remove sites with ambiguity data (1:yes, 0:no)?
BDparas = 1 1 0 * birth, death, sampling
kappa_gamma = 6 2 * gamma prior for kappa
alpha_gamma = 1 1 * gamma prior for alpha
rgene_gamma = 2 2 * gamma prior for overall rates for genes
sigma2_gamma = 1 10 * gamma prior for sigma^2 (for clock=2 or 3)
finetune = 1: 0.1 0.1 0.1 0.01 .5 * auto (0 or 1) : times, musigma2, rates, mixing, paras, FossilErr
print = 1
burnin = 2000
sampfreq = 2
nsample = 20000
*** Note: Make your window wider (100 columns) before running the program.
thank to this tuorial to guide me throught the output management
http://abacus.gene.ucl.ac.uk/software/MCMCtree.Tutorials.pdf
and
http://www.jcsantosresearch.org/Class_2014_Spring_Comparative/pdf/week_6/Feb_12_2015_Chronograms.pdf
To gat this I firstly use
24 July 2021
My topic of the day was how to extract intron and intergenic specaer for the phylogenetic tree construction?
I got a good discussion on that here or here. It is retained that GFF-ex is very nice to do that job. Get the tool here. How to install and run here
I will give a try soon.
wget http://bioinfo.icgeb.res.in/gff/gffdownloads/GFF_v2.3.tar.gz
gunzip
tar -vxf
[yedomon@localhost utils]$ cd GFF_v2.3/
[yedomon@localhost GFF_v2.3]$ ll
total 16
drwxrwxr-x. 2 yedomon yedomon 90 Oct 13 2014 examples
drwxrwxr-x. 2 yedomon yedomon 4096 Jan 13 2016 gff_scripts
-rwxrwxr-x. 1 yedomon yedomon 581 Jan 13 2016 gff_usage.txt
-rwxrwxr-x. 1 yedomon yedomon 1109 Jan 13 2016 install.sh
-rwxrwxr-x. 1 yedomon yedomon 3288 Jan 13 2016 README
[yedomon@localhost GFF_v2.3]$ ./install.sh /home/yedomon/utils/GFF_v2.3/
cp: ‘/home/yedomon/utils/GFF_v2.3/gff_scripts/’ and ‘/home/yedomon/utils/GFF_v2.3/gff_scripts’ are the same file
[SUCCESS]:GFF-Ex installation path set to /home/yedomon/utils/GFF_v2.3
[SUCCESS]:GFF-Ex. files copied to /home/yedomon/utils/GFF_v2.3
[PERFORM]:To use the program, Run -> source /home/yedomon/utils/GFF_v2.3/gff_scripts/gff_profile
[PERFORM]:Once above command is executed, using same terminal, move to directoty containing your input files and run GFF-Ex as [/home/yedomon/utils/GFF_v2.3/gff_scripts/gffex -in <gfffile> -db <genomefile>]
[NOTE]:Please remember to run [source /home/yedomon/utils/GFF_v2.3/gff_scripts/gff_profile] everytime, before executing gffex
[yedomon@localhost GFF_v2.3]$
But the issu the is no gff_profile in that folder /home/yedomon/utils/GFF_v2.3/ so i tried again
[yedomon@localhost GFF_v2.3]$ ./install.sh /home/yedomon/utils/GFF_v2.3/gff_scripts
copying scripts to /home/yedomon/utils/GFF_v2.3/gff_scripts/
cp: cannot copy a directory, ‘/home/yedomon/utils/GFF_v2.3/gff_scripts/’, into itself, ‘/home/yedomon/utils/GFF_v2.3/gff_scripts/gff_scripts’
[SUCCESS]:GFF-Ex installation path set to /home/yedomon/utils/GFF_v2.3/gff_scripts
[SUCCESS]:GFF-Ex. files copied to /home/yedomon/utils/GFF_v2.3/gff_scripts
[PERFORM]:To use the program, Run -> source /home/yedomon/utils/GFF_v2.3/gff_scripts/gff_scripts/gff_profile
[PERFORM]:Once above command is executed, using same terminal, move to directoty containing your input files and run GFF-Ex as [/home/yedomon/utils/GFF_v2.3/gff_scripts/gff_scripts/gffex -in <gfffile> -db <genomefile>]
[NOTE]:Please remember to run [source /home/yedomon/utils/GFF_v2.3/gff_scripts/gff_scripts/gff_profile] everytime, before executing gffex
source /home/yedomon/utils/GFF_v2.3/gff_scripts/gff_profile
Test
/home/yedomon/utils/GFF_v2.3/gff_scripts/gff_scripts/gffex \
-in /home/yedomon/data/01_ka_ks/01_sesamum_indicum_goenbaek/goenbaek_GeSeqJob-20210700-161938_Sesamum_indicum_cv_Goenbaek_GFF3.gff3 \
-db /home/yedomon/data/01_ka_ks/01_sesamum_indicum_goenbaek/Sesamum_indicum_cv_Goenbaek.fasta
blem de gff3 gff2 or gtf je crois.
Ici on me propoe une soluition https://www.biostars.org/p/112251/
# step1
awk 'OFS="\t" {print $1, "0", $2}' chromSizes.txt | sort -k1,1 -k2,2n > chromSizes.bed
# step2
cat in.gff | awk '$1 ~ /^#/ {print $0;next} {print $0 | "sort -k1,1 -k4,4n -k5,5n"}' > in_sorted.gff
# get intergenic region
bedtools complement -i in_sorted.gff -g chromSizes.txt > intergenic_sorted.bed
# get intron
awk 'OFS="\t", $1 ~ /^#/ {print $0;next} {if ($3 == "exon") print $1, $4-1, $5}' in_sorted.gff > exon_sorted.bed
bedtools complement -i <(cat exon_sorted.bed intergenic_sorted.bed | sort -k1,1 -k2,2n) -g chromSizes.txt > intron_sorted.bed
un autre https://davetang.org/muse/2013/01/18/defining-genomic-regions/
un autre http://crazyhottommy.blogspot.com/2013/05/find-exons-introns-and-intergenic.html
un autre https://blog.daum.net/naturelove87/179
un autre http://quinlanlab.org/tutorials/bedtools/bedtools.html
bedtools complement -i exons.bed -g genome.txt \
> non-exonic.bed
head non-exonic.bed
chr1 0 11873
chr1 12227 12612
chr1 12721 13220
chr1 14829 14969
chr1 15038 15795
chr1 15947 16606
chr1 16765 16857
chr1 17055 17232
chr1 17368 17605
chr1 17742 17914
je vais utiliser ceci voir https://www.reneshbedre.com/blog/gffgtf.html via conda
Another topic is time divergence estimation
Step 1: Do a phylogenetic tree
In my case a have both Bayesian and ML tree based on CDS
Step 2: Find a boundaries times for calibration
Critical point. For my case, I will introduce arabidopsis outgroup , Echinacanthus prendre les trois Closer family Acantacae en bas ) and les trois Torenia (Linderniacae closer famuily en haut). to find the different calibration use timetree.org two species and enter the corresponding pairwise genus to get the CI that will be incorporated in the netwick tree
The time unit in the analysis is 100 million years (My). Thus, the calibration B(0.075, 0.10) means the node age is constrained to be between 7.5 and 10 million years ago (Ma)
from these tuto rule 1 : no branch length, rule 2 use ATGC alignment instead of protein .
Chloroplast available
Step 3:
Just found today 23 july 2021 a tool for plotting phylogenetic tree
github | Time divergence | Drawing taxon images | Drawing an alignment with the tree
ALiTV installation for chloroplast genome comparison
I went to the ALITV github page for checking.
External Requirements:
perl
cpanm
lastz
So let's install those requirements first.
# Install perl and cpanm
sudo yum install perl-devel
sudo yum install cpanminus
sudo cpanm Bio::Perl # to install bioperl tools
sudo cpanm --force Bio::Perl # force failed installation module
# install lastz
## Download source code
wget https://github.com/AliTVTeam/AliTV-perl-interface/archive/refs/heads/master.zip
## Unzip it
unzip lastz-master.zip # I did it on windows and there was a double folders inside (latsz-master/lastz-master/files) so I just grab the second lastz folder
## Setting the installation path
LASTZ_INSTALL=/home/yedomon/utils/lastz-master/src
## Open the .bashrc
cd $HOME
vi .bashrc
## write
export PATH=/home/yedomon/utils/lastz-master/src:$PATH or export PATH=$PATH:/place/with/the/file
## DO
Esc + : + wq
## Then
source .bashrc
## Then
cd /home/yedomon/utils/lastz-master/src
make
make install
the log file of make and make install
[yedomon@localhost src]$ make
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' lastz.c -o lastz.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' infer_scores.c -o infer_scores.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' seeds.c -o seeds.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' pos_table.c -o pos_table.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' quantum.c -o quantum.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' seed_search.c -o seed_search.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' diag_hash.c -o diag_hash.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' chain.c -o chain.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' gapped_extend.c -o gapped_extend.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' tweener.c -o tweener.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' masking.c -o masking.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' segment.c -o segment.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' edit_script.c -o edit_script.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' identity_dist.c -o identity_dist.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' coverage_dist.c -o coverage_dist.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' continuity_dist.c -o continuity_dist.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' output.c -o output.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' gfa.c -o gfa.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' lav.c -o lav.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' axt.c -o axt.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' maf.c -o maf.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' cigar.c -o cigar.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' sam.c -o sam.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' genpaf.c -o genpaf.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' text_align.c -o text_align.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' align_diffs.c -o align_diffs.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' utilities.c -o utilities.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' dna_utilities.c -o dna_utilities.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' sequences.c -o sequences.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'I\' capsule.c -o capsule.o
gcc lastz.o infer_scores.o seeds.o pos_table.o quantum.o seed_search.o diag_hash.o chain.o gapped_extend.o tweener.o masking.o segment.o edit_script.o identity_dist.o coverage_dist.o continuity_dist.o output.o gfa.o lav.o axt.o maf.o cigar.o sam.o genpaf.o text_align.o align_diffs.o utilities.o dna_utilities.o sequences.o capsule.o -lm -o lastz
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' lastz.c -o lastz_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' infer_scores.c -o infer_scores_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' seeds.c -o seeds_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' pos_table.c -o pos_table_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' quantum.c -o quantum_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' seed_search.c -o seed_search_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' diag_hash.c -o diag_hash_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' chain.c -o chain_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' gapped_extend.c -o gapped_extend_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' tweener.c -o tweener_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' masking.c -o masking_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' segment.c -o segment_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' edit_script.c -o edit_script_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' identity_dist.c -o identity_dist_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' coverage_dist.c -o coverage_dist_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' continuity_dist.c -o continuity_dist_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' output.c -o output_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' gfa.c -o gfa_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' lav.c -o lav_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' axt.c -o axt_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' maf.c -o maf_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' cigar.c -o cigar_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' sam.c -o sam_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' genpaf.c -o genpaf_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' text_align.c -o text_align_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' align_diffs.c -o align_diffs_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' utilities.c -o utilities_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' dna_utilities.c -o dna_utilities_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' sequences.c -o sequences_D.o
gcc -c -O3 -Wall -Wextra -Werror -D_FILE_OFFSET_BITS=64 -D_LARGEFILE_SOURCE -DVERSION_MAJOR="\"1"\" -DVERSION_MINOR="\"04"\" -DVERSION_SUBMINOR="\"12"\" -DREVISION_DATE="\"20210209"\" -DSUBVERSION_REV="\""\" -Dscore_type=\'D\' capsule.c -o capsule_D.o
gcc lastz_D.o infer_scores_D.o seeds_D.o pos_table_D.o quantum_D.o seed_search_D.o diag_hash_D.o chain_D.o gapped_extend_D.o tweener_D.o masking_D.o segment_D.o edit_script_D.o identity_dist_D.o coverage_dist_D.o continuity_dist_D.o output_D.o gfa_D.o lav_D.o axt_D.o maf_D.o cigar_D.o sam_D.o genpaf_D.o text_align_D.o align_diffs_D.o utilities_D.o dna_utilities_D.o sequences_D.o capsule_D.o -lm -o lastz_D
[yedomon@localhost src]$ make install
install -d /home/yedomon/lastz-distrib/bin
install lastz /home/yedomon/lastz-distrib/bin
install lastz_D /home/yedomon/lastz-distrib/bin
[yedomon@localhost src]$
Then
make testOutput
[yedomon@localhost src]$ make test
[yedomon@localhost src]$
No error message came. So That is good. LASTZ is well installed.
Now Let's Install AliTv
cd /home/yedomon/utils # Move to my software installation directory
git clone https://github.com/AliTVTeam/AliTV-perl-interface
cd AliTV-perl-interface/bin/
# Both executables are there
# -rwxrwxr-x. 1 yedomon yedomon 11355 Jul 22 10:18 alitv-filter.pl
# -rwxrwxr-x. 1 yedomon yedomon 7284 Jul 22 10:18 alitv.pl
# I will try to run it
```python
[yedomon@localhost bin]$ ./alitv.pl --help
Can't locate FindBin/Real.pm in @INC (@INC contains: /usr/local/lib64/perl5 /usr/local/share/perl5 /usr/lib64/perl5/vendor_perl /usr/share/perl5/vendor_perl /usr/lib64/perl5 /usr/share/perl5 .) at ./alitv.pl line 6.
BEGIN failed--compilation aborted at ./alitv.pl line 6.So I went back
cd ../../
# Then
[yedomon@localhost utils]$ cpanm --installdeps .
!
! Can't write to /usr/local/share/perl5 and /usr/local/bin: Installing modules to /home/yedomon/perl5
! To turn off this warning, you have to do one of the following:
! - run me as a root or with --sudo option (to install to /usr/local/share/perl5 and /usr/local/bin)
! - Configure local::lib your existing local::lib in this shell to set PERL_MM_OPT etc.
! - Install local::lib by running the following commands
!
! cpanm --local-lib=~/perl5 local::lib && eval $(perl -I ~/perl5/lib/perl5/ -Mlocal::lib)
!
--> Working on .
Configuring /home/yedomon/utils ... N/A
! Configuring . failed. See /home/yedomon/.cpanm/work/1626917121.21810/build.log for details.
# I use sudo
[yedomon@localhost utils]$ sudo cpanm --installdeps .
[sudo] password for yedomon:
--> Working on .
Configuring /home/yedomon/utils ... N/A
! Configuring . failed. See /root/.cpanm/work/1626917196.22270/build.log for details.
# Then I use this
[yedomon@localhost utils]$ cpanm --local-lib=~/perl5 local::lib && eval $(perl -I ~/perl5/lib/perl5/ -Mlocal::lib)
--> Working on local::lib
Fetching http://www.cpan.org/authors/id/H/HA/HAARG/local-lib-2.000024.tar.gz ... OK
Configuring local-lib-2.000024 ... OK
==> Found dependencies: ExtUtils::MakeMaker
--> Working on ExtUtils::MakeMaker
Fetching http://www.cpan.org/authors/id/B/BI/BINGOS/ExtUtils-MakeMaker-7.62.tar.gz ... OK
Configuring ExtUtils-MakeMaker-7.62 ... OK
Building and testing ExtUtils-MakeMaker-7.62 ... OK
Successfully installed ExtUtils-MakeMaker-7.62 (upgraded from 6.68)
Building and testing local-lib-2.000024 ... OK
Successfully installed local-lib-2.000024
2 distributions installed
Attempting to create directory /home/yedomon/perl5
[yedomon@localhost utils]$
Then I run
[yedomon@localhost bin]$ perl alitv.pl --help
Can't locate FindBin/Real.pm in @INC (@INC contains: /home/yedomon/perl5/lib/perl5/5.16.3/x86_64-linux-thread-multi /home/yedomon/perl5/lib/perl5/5.16.3 /home/yedomon/perl5/lib/perl5/x86_64-linux-thread-multi /home/yedomon/perl5/lib/perl5 /usr/local/lib64/perl5 /usr/local/share/perl5 /usr/lib64/perl5/vendor_perl /usr/share/perl5/vendor_perl /usr/lib64/perl5 /usr/share/perl5 .) at alitv.pl line 6.
BEGIN failed--compilation aborted at alitv.pl line 6.
[yedomon@localhost bin]$ sudo cpanm install FindBin::Real
[sudo] password for yedomon:
--> Working on install
Fetching http://www.cpan.org/authors/id/D/DA/DAGOLDEN/install-0.01.tar.gz ... OK
Configuring install-0.01 ... OK
Building and testing install-0.01 ... OK
Successfully installed install-0.01
--> Working on FindBin::Real
Fetching http://www.cpan.org/authors/id/S/ST/STRO/FindBin-Real-1.05.tar.gz ... OK
Configuring FindBin-Real-1.05 ... OK
Building and testing FindBin-Real-1.05 ... OK
Successfully installed FindBin-Real-1.05
2 distributions installed
[yedomon@localhost bin]$ perl alitv.pl --help
Can't locate Log/Log4perl.pm in @INC (@INC contains: /home/yedomon/utils/AliTV-perl-interface/bin/../lib /home/yedomon/perl5/lib/perl5/5.16.3/x86_64-linux-thread-multi /home/yedomon/perl5/lib/perl5/5.16.3 /home/yedomon/perl5/lib/perl5/x86_64-linux-thread-multi /home/yedomon/perl5/lib/perl5 /usr/local/lib64/perl5 /usr/local/share/perl5 /usr/lib64/perl5/vendor_perl /usr/share/perl5/vendor_perl /usr/lib64/perl5 /usr/share/perl5 .) at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV/Base.pm line 4.
BEGIN failed--compilation aborted at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV/Base.pm line 4.
Compilation failed in require at /usr/share/perl5/vendor_perl/parent.pm line 20.
BEGIN failed--compilation aborted at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV.pm line 7.
Compilation failed in require at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV/Script.pm line 9.
BEGIN failed--compilation aborted at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV/Script.pm line 9.
Compilation failed in require at alitv.pl line 9.
BEGIN failed--compilation aborted at alitv.pl line 9.
[yedomon@localhost bin]$ sudo cpanm install Log::Log4perl
install is up to date. (0.01)
--> Working on Log::Log4perl
Fetching http://www.cpan.org/authors/id/E/ET/ETJ/Log-Log4perl-1.54.tar.gz ... OK
Configuring Log-Log4perl-1.54 ... OK
Building and testing Log-Log4perl-1.54 ... OK
Successfully installed Log-Log4perl-1.54
1 distribution installed
[yedomon@localhost bin]$ perl alitv.pl --help
Can't locate Hash/Merge.pm in @INC (@INC contains: /home/yedomon/utils/AliTV-perl-interface/bin/../lib /home/yedomon/perl5/lib/perl5/5.16.3/x86_64-linux-thread-multi /home/yedomon/perl5/lib/perl5/5.16.3 /home/yedomon/perl5/lib/perl5/x86_64-linux-thread-multi /home/yedomon/perl5/lib/perl5 /usr/local/lib64/perl5 /usr/local/share/perl5 /usr/lib64/perl5/vendor_perl /usr/share/perl5/vendor_perl /usr/lib64/perl5 /usr/share/perl5 .) at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV.pm line 12.
BEGIN failed--compilation aborted at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV.pm line 12.
Compilation failed in require at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV/Script.pm line 9.
BEGIN failed--compilation aborted at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV/Script.pm line 9.
Compilation failed in require at alitv.pl line 9.
BEGIN failed--compilation aborted at alitv.pl line 9.
[yedomon@localhost bin]$ sudo cpanm install Hash::Merge
install is up to date. (0.01)
--> Working on Hash::Merge
Fetching http://www.cpan.org/authors/id/H/HE/HERMES/Hash-Merge-0.302.tar.gz ... OK
Configuring Hash-Merge-0.302 ... OK
==> Found dependencies: Clone::Choose
--> Working on Clone::Choose
Fetching http://www.cpan.org/authors/id/H/HE/HERMES/Clone-Choose-0.010.tar.gz ... OK
Configuring Clone-Choose-0.010 ... OK
==> Found dependencies: Test::Without::Module
--> Working on Test::Without::Module
Fetching http://www.cpan.org/authors/id/C/CO/CORION/Test-Without-Module-0.20.tar.gz ... OK
Configuring Test-Without-Module-0.20 ... OK
Building and testing Test-Without-Module-0.20 ... OK
Successfully installed Test-Without-Module-0.20
Building and testing Clone-Choose-0.010 ... OK
Successfully installed Clone-Choose-0.010
Building and testing Hash-Merge-0.302 ... OK
Successfully installed Hash-Merge-0.302
3 distributions installed
[yedomon@localhost bin]$ perl alitv.pl --help
Can't locate JSON.pm in @INC (@INC contains: /home/yedomon/utils/AliTV-perl-interface/bin/../lib /home/yedomon/perl5/lib/perl5/5.16.3/x86_64-linux-thread-multi /home/yedomon/perl5/lib/perl5/5.16.3 /home/yedomon/perl5/lib/perl5/x86_64-linux-thread-multi /home/yedomon/perl5/lib/perl5 /usr/local/lib64/perl5 /usr/local/share/perl5 /usr/lib64/perl5/vendor_perl /usr/share/perl5/vendor_perl /usr/lib64/perl5 /usr/share/perl5 .) at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV.pm line 19.
BEGIN failed--compilation aborted at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV.pm line 19.
Compilation failed in require at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV/Script.pm line 9.
BEGIN failed--compilation aborted at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV/Script.pm line 9.
Compilation failed in require at alitv.pl line 9.
BEGIN failed--compilation aborted at alitv.pl line 9.
[yedomon@localhost bin]$ sudo cpanm install JSON
install is up to date. (0.01)
--> Working on JSON
Fetching http://www.cpan.org/authors/id/I/IS/ISHIGAKI/JSON-4.03.tar.gz ... OK
Configuring JSON-4.03 ... OK
Building and testing JSON-4.03 ... OK
Successfully installed JSON-4.03
1 distribution installed
[yedomon@localhost bin]$ perl alitv.pl --help
Usage:
# complex configuration via yml file
alitv.pl [OPTIONS] options.yml
# OR
# easy alternative including the generation of a yml file
alitv.pl [OPTIONS] *.fasta
Options:
--project Project name
The name of the project will be the given argument. If this
parameter was not provided, one project name will be auto generated.
This will be the base name for the log file, the yml file, and the
output file. If a YML file is provided, this value will be
overwritten by the basename of the YML file.
--output Output file
The name of the output file. If non is provided, the output file
name will be based on the project name. If STDOUT should be used,
please set the output filename to "-" via option "alitv.pl --output
-".
--logfile Log file
The name of the log file. If non is provided, the log file name will
be based on the project name.
--overwrite or --force Overwrite existing project.yml or output.json
files
Default behaviour is to keep existing project yml and json files. If
"--overwrite" or "--force" is specified, the files will be
overwritten. Overwriting can be expicitly disabled by
"--no-overwrite" or "--no-force" parameter.
In summary all those error meassage were due to the missing package, FindBin/Real, log/Log4perl, Hash/Merge and JSON So to solve the problem, I did
sudo cpanm install FindBin::Real
sudo cpanm install Log::Log4perl
sudo cpanm install Hash::Merge
sudo cpanm install JSON
sudo cpanm install YAML
# and
sudo cpanm --installdeps .Then It works fine!
Now I can run the tool with my own data
First I will create a folder containing all fasta, genbank files and tree file in netwick format
cd /home/yedomon/utils/AliTV-perl-interface/bin/
perl alitv.pl
--project set9
/home/yedomon/utils/AliTV-perl-interface/data/chloro9/*.fasta
export PATH=/home/yedomon/utils/AliTV-perl-interface/bin:$PATH
perl bin/alitv.pl --project set9 --overwrite data/chloro9/set9.yml
[yedomon@localhost AliTV-perl-interface]$ perl bin/alitv.pl --project set9 --overwrite data/chloro9/set9.yml
***********************************************************************
* *
* AliTV perl interface *
* *
***********************************************************************
You are using version v1.0.6.
INFO - Sequence names are longer then maximum allowed length (8 characters) and will be replaced by unique sequence names. Failing sequence names are: 'Ceratotheca_sesamoides', 'Ceratotheca_triloba', 'Sesamum_alatum', 'Sesamum_angolense', 'Sesamum_indicum_cv_Ansanggae', 'Sesamum_indicum_cv_Goenbaek', 'Sesamum_indicum_cv_Yuzhi11', 'Sesamum_pedaloides', 'Sesamum_radiatum'
FATAL - Unable to load alignment module 'AliTV::Alignment::lastz' at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV/Script.pm line 123.
Unable to load alignment module 'AliTV::Alignment::lastz' at /home/yedomon/utils/AliTV-perl-interface/bin/../lib/AliTV/Script.pm line 123.
[yedomon@localhost AliTV-perl-interface]$ sudo cpanm --installdeps .
[sudo] password for yedomon:
--> Working on .
Configuring /home/yedomon/utils/AliTV-perl-interface ... OK
==> Found dependencies: Bio::FeatureIO, Test::Warnings, Test::Exit, IPC::System::Simple
--> Working on Bio::FeatureIO
Fetching http://www.cpan.org/authors/id/C/CJ/CJFIELDS/Bio-FeatureIO-1.6.905.tar.gz ... OK
Configuring Bio-FeatureIO-1.6.905 ... OK
==> Found dependencies: XML::DOM::XPath, Tree::DAG_Node
--> Working on XML::DOM::XPath
Fetching http://www.cpan.org/authors/id/M/MI/MIROD/XML-DOM-XPath-0.14.tar.gz ... OK
Configuring XML-DOM-XPath-0.14 ... OK
==> Found dependencies: XML::XPathEngine
--> Working on XML::XPathEngine
Fetching http://www.cpan.org/authors/id/M/MI/MIROD/XML-XPathEngine-0.14.tar.gz ... OK
Configuring XML-XPathEngine-0.14 ... OK
Building and testing XML-XPathEngine-0.14 ... OK
Successfully installed XML-XPathEngine-0.14
Building and testing XML-DOM-XPath-0.14 ... OK
Successfully installed XML-DOM-XPath-0.14
--> Working on Tree::DAG_Node
Fetching http://www.cpan.org/authors/id/R/RS/RSAVAGE/Tree-DAG_Node-1.32.tgz ... OK
Configuring Tree-DAG_Node-1.32 ... OK
==> Found dependencies: File::Slurp::Tiny
--> Working on File::Slurp::Tiny
Fetching http://www.cpan.org/authors/id/L/LE/LEONT/File-Slurp-Tiny-0.004.tar.gz ... OK
Configuring File-Slurp-Tiny-0.004 ... OK
Building and testing File-Slurp-Tiny-0.004 ... OK
Successfully installed File-Slurp-Tiny-0.004
Building and testing Tree-DAG_Node-1.32 ... OK
Successfully installed Tree-DAG_Node-1.32
Building and testing Bio-FeatureIO-1.6.905 ... OK
Successfully installed Bio-FeatureIO-1.6.905
--> Working on Test::Warnings
Fetching http://www.cpan.org/authors/id/E/ET/ETHER/Test-Warnings-0.031.tar.gz ... OK
Configuring Test-Warnings-0.031 ... OK
Building and testing Test-Warnings-0.031 ... OK
Successfully installed Test-Warnings-0.031
--> Working on Test::Exit
Fetching http://www.cpan.org/authors/id/A/AR/ARODLAND/Test-Exit-0.11.tar.gz ... OK
Configuring Test-Exit-0.11 ... OK
==> Found dependencies: Return::MultiLevel
--> Working on Return::MultiLevel
Fetching http://www.cpan.org/authors/id/M/MA/MAUKE/Return-MultiLevel-0.05.tar.gz ... OK
Configuring Return-MultiLevel-0.05 ... OK
==> Found dependencies: Data::Munge
--> Working on Data::Munge
Fetching http://www.cpan.org/authors/id/M/MA/MAUKE/Data-Munge-0.097.tar.gz ... OK
Configuring Data-Munge-0.097 ... OK
Building and testing Data-Munge-0.097 ... OK
Successfully installed Data-Munge-0.097
Building and testing Return-MultiLevel-0.05 ... FAIL
! Installing Return::MultiLevel failed. See /root/.cpanm/work/1626926105.16802/build.log for details. Retry with --force to force install it.
! Installing the dependencies failed: Module 'Return::MultiLevel' is not installed
! Bailing out the installation for Test-Exit-0.11.
--> Working on IPC::System::Simple
Fetching http://www.cpan.org/authors/id/J/JK/JKEENAN/IPC-System-Simple-1.30.tar.gz ... OK
Configuring IPC-System-Simple-1.30 ... OK
Building and testing IPC-System-Simple-1.30 ... OK
Successfully installed IPC-System-Simple-1.30
! Installing the dependencies failed: Module 'Test::Exit' is not installed
! Bailing out the installation for AliTV-v1.0.6.
8 distributions installed
[yedomon@localhost AliTV-perl-interface]$ sudo cpanm --force --installdeps .
--> Working on .
Configuring /home/yedomon/utils/AliTV-perl-interface ... OK
==> Found dependencies: Test::Exit
--> Working on Test::Exit
Fetching http://www.cpan.org/authors/id/A/AR/ARODLAND/Test-Exit-0.11.tar.gz ... OK
Configuring Test-Exit-0.11 ... OK
==> Found dependencies: Return::MultiLevel
--> Working on Return::MultiLevel
Fetching http://www.cpan.org/authors/id/M/MA/MAUKE/Return-MultiLevel-0.05.tar.gz ... OK
Configuring Return-MultiLevel-0.05 ... OK
Building and testing Return-MultiLevel-0.05 ... FAIL
! Testing Return-MultiLevel-0.05 failed but installing it anyway.
Successfully installed Return-MultiLevel-0.05
Building and testing Test-Exit-0.11 ... OK
Successfully installed Test-Exit-0.11
<== Installed dependencies for .. Finishing.
2 distributions installed
[yedomon@localhost AliTV-perl-interface]$ perl bin/alitv.pl --project set9 --overwrite data/chloro9/set9.yml
***********************************************************************
* *
* AliTV perl interface *
* *
***********************************************************************
You are using version v1.0.6.
INFO - Sequence names are longer then maximum allowed length (8 characters) and will be replaced by unique sequence names. Failing sequence names are: 'Ceratotheca_sesamoides', 'Ceratotheca_triloba', 'Sesamum_alatum', 'Sesamum_angolense', 'Sesamum_indicum_cv_Ansanggae', 'Sesamum_indicum_cv_Goenbaek', 'Sesamum_indicum_cv_Yuzhi11', 'Sesamum_pedaloides', 'Sesamum_radiatum'
INFO - Created temporary folder at '/tmp/i9SBadbWJC'
INFO - Starting alignment generation... (45 alignments required)
INFO - Finished 1. alignment (44 to go; 2.22 % done)
INFO - Finished 2. alignment (43 to go; 4.44 % done)
INFO - Finished 3. alignment (42 to go; 6.67 % done)
INFO - Finished 4. alignment (41 to go; 8.89 % done)
INFO - Finished 5. alignment (40 to go; 11.11 % done)
INFO - Finished 6. alignment (39 to go; 13.33 % done)
INFO - Finished 7. alignment (38 to go; 15.56 % done)
INFO - Finished 8. alignment (37 to go; 17.78 % done)
INFO - Finished 9. alignment (36 to go; 20.00 % done)
INFO - Finished 10. alignment (35 to go; 22.22 % done)
INFO - Finished 11. alignment (34 to go; 24.44 % done)
INFO - Finished 12. alignment (33 to go; 26.67 % done)
INFO - Finished 13. alignment (32 to go; 28.89 % done)
INFO - Finished 14. alignment (31 to go; 31.11 % done)
INFO - Finished 15. alignment (30 to go; 33.33 % done)
INFO - Finished 16. alignment (29 to go; 35.56 % done)
INFO - Finished 17. alignment (28 to go; 37.78 % done)
INFO - Finished 18. alignment (27 to go; 40.00 % done)
INFO - Finished 19. alignment (26 to go; 42.22 % done)
INFO - Finished 20. alignment (25 to go; 44.44 % done)
INFO - Finished 21. alignment (24 to go; 46.67 % done)
INFO - Finished 22. alignment (23 to go; 48.89 % done)
INFO - Finished 23. alignment (22 to go; 51.11 % done)
INFO - Finished 24. alignment (21 to go; 53.33 % done)
INFO - Finished 25. alignment (20 to go; 55.56 % done)
INFO - Finished 26. alignment (19 to go; 57.78 % done)
INFO - Finished 27. alignment (18 to go; 60.00 % done)
INFO - Finished 28. alignment (17 to go; 62.22 % done)
INFO - Finished 29. alignment (16 to go; 64.44 % done)
INFO - Finished 30. alignment (15 to go; 66.67 % done)
INFO - Finished 31. alignment (14 to go; 68.89 % done)
INFO - Finished 32. alignment (13 to go; 71.11 % done)
INFO - Finished 33. alignment (12 to go; 73.33 % done)
INFO - Finished 34. alignment (11 to go; 75.56 % done)
INFO - Finished 35. alignment (10 to go; 77.78 % done)
INFO - Finished 36. alignment (9 to go; 80.00 % done)
INFO - Finished 37. alignment (8 to go; 82.22 % done)
INFO - Finished 38. alignment (7 to go; 84.44 % done)
INFO - Finished 39. alignment (6 to go; 86.67 % done)
INFO - Finished 40. alignment (5 to go; 88.89 % done)
INFO - Finished 41. alignment (4 to go; 91.11 % done)
INFO - Finished 42. alignment (3 to go; 93.33 % done)
INFO - Finished 43. alignment (2 to go; 95.56 % done)
INFO - Finished 44. alignment (1 to go; 97.78 % done)
INFO - Finished 45. alignment (0 to go; 100.00 % done)
INFO - Finished alignment generation
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - Deleting temporary folder
INFO - Number of bases (1378837) is longer than the maximum allowed (1000000), therefore sequences will be excluded from JSON file
INFO - Ticks will be drawn every 1000 basepair
Using 9 species is too much. Maximum is 8.
So I just keep one sesamum indicum | The reference one from Korea.
I will do the tree. So I need to prepare the files
[yedomon@localhost set_1]$ cp *tri* ../set_7
[yedomon@localhost set_1]$ cp *cse* ../set_7
[yedomon@localhost set_1]$ cp *ans* ../set_7
[yedomon@localhost set_1]$ cp *ang* ../set_7
[yedomon@localhost set_1]$ cp *ped* ../set_7
[yedomon@localhost set_1]$ cp *rad* ../set_7
[yedomon@localhost set_1]$ cp *ala* ../set_7
[yedomon@localhost set_1]$ cd ../set_7
Then make the tree
#/bin/bash
set -e
cd /NABIC/HOME/yedomon1/plastomics/20.phylo/set7
for i in *.faa
do
### Multiple sequence alignment
source activate mafft_env
mafft --thread 32 --auto $i > $i.mafft
source deactivate mafft_env
### Alignment trimming
source activate trimal_env
trimal -automated1 -in $i.mafft -out $i.mafft.trimal
source deactivate trimal_env
done
##---Make the matrix
cat *.mafft.trimal | awk -v RS=">" -v FS="\n" -v OFS="\n" '{for(i=2; i<=NF; i++) {seq[$1] = seq[$1]$i}}; END {for(id in seq){print ">"id, seq[id]}}' > combined.awk.fasta
mkdir tree_construction
cd tree_construction
cp ../combined.awk.fasta .
source activate iqtree_env
iqtree -s combined.awk.fasta -nt AUTO -bb 1000 -alrt 1000
source deactivate iqtree_env
## Bye!
bash run_phylo.sh &> log.phylo &
VERY IMPORTANT Hint: To include the tree in AliTv the name of the chloroplast sequence should match those of the tree
So I prepare my input.yml file and chloroplast data in one folder
My yaml file is like this
---
alignment:
parameter:
- --format=maf
- --noytrim
- --ambiguous=iupac
- --gapped
- --strand=both
program: lastz
genomes:
- name: Ceratotheca_sesamoides
sequence_files:
- data/chloro7/Ceratotheca_sesamoides.fasta
- name: Ceratotheca_triloba
sequence_files:
- data/chloro7/Ceratotheca_triloba.fasta
- name: Sesamum_alatum
sequence_files:
- data/chloro7/Sesamum_alatum.fasta
- name: Sesamum_angolense
sequence_files:
- data/chloro7/Sesamum_angolense.fasta
- name: Sesamum_indicum_cv_Ansanggae
sequence_files:
- data/chloro7/Sesamum_indicum_cv_Ansanggae.fasta
- name: Sesamum_pedaloides
sequence_files:
- data/chloro7/Sesamum_pedaloides.fasta
- name: Sesamum_radiatum
sequence_files:
- data/chloro7/Sesamum_radiatum.fasta
tree: data/chloro7/species.tree
then run
cd /home/yedomon/utils/AliTV-perl-interface # positioning accordingly ....very important ...check the yaml file..That matches perfectely. AliTv can find the files
perl bin/alitv.pl --project set7 data/chloro7/set7.yml
log
***********************************************************************
* *
* AliTV perl interface *
* *
***********************************************************************
You are using version v1.0.6.
INFO - Sequence names are longer then maximum allowed length (8 characters) and will be replaced by unique sequence names. Failing sequence names are: 'Ceratotheca_sesamoides', 'Ceratotheca_triloba', 'Sesamum_alatum', 'Sesamum_angolense', 'Sesamum_indicum_cv_Ansanggae', 'Sesamum_pedaloides', 'Sesamum_radiatum'
INFO - Created temporary folder at '/tmp/HxNqI4umFD'
INFO - Starting alignment generation... (28 alignments required)
INFO - Finished 1. alignment (27 to go; 3.57 % done)
INFO - Finished 2. alignment (26 to go; 7.14 % done)
INFO - Finished 3. alignment (25 to go; 10.71 % done)
INFO - Finished 4. alignment (24 to go; 14.29 % done)
INFO - Finished 5. alignment (23 to go; 17.86 % done)
INFO - Finished 6. alignment (22 to go; 21.43 % done)
INFO - Finished 7. alignment (21 to go; 25.00 % done)
INFO - Finished 8. alignment (20 to go; 28.57 % done)
INFO - Finished 9. alignment (19 to go; 32.14 % done)
INFO - Finished 10. alignment (18 to go; 35.71 % done)
INFO - Finished 11. alignment (17 to go; 39.29 % done)
INFO - Finished 12. alignment (16 to go; 42.86 % done)
INFO - Finished 13. alignment (15 to go; 46.43 % done)
INFO - Finished 14. alignment (14 to go; 50.00 % done)
INFO - Finished 15. alignment (13 to go; 53.57 % done)
INFO - Finished 16. alignment (12 to go; 57.14 % done)
INFO - Finished 17. alignment (11 to go; 60.71 % done)
INFO - Finished 18. alignment (10 to go; 64.29 % done)
INFO - Finished 19. alignment (9 to go; 67.86 % done)
INFO - Finished 20. alignment (8 to go; 71.43 % done)
INFO - Finished 21. alignment (7 to go; 75.00 % done)
INFO - Finished 22. alignment (6 to go; 78.57 % done)
INFO - Finished 23. alignment (5 to go; 82.14 % done)
INFO - Finished 24. alignment (4 to go; 85.71 % done)
INFO - Finished 25. alignment (3 to go; 89.29 % done)
INFO - Finished 26. alignment (2 to go; 92.86 % done)
INFO - Finished 27. alignment (1 to go; 96.43 % done)
INFO - Finished 28. alignment (0 to go; 100.00 % done)
INFO - Finished alignment generation
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - MAF input file detected, but Bioperl is bugfree... Therefore, workaround for revcom issue is not activated
INFO - Deleting temporary folder
INFO - Number of bases (1072161) is longer than the maximum allowed (1000000), therefore sequences will be excluded from JSON file
INFO - Ticks will be drawn every 1000 basepair
[yedomon@localhost AliTV-perl-interface]$
I upload but it was empty. I tried the default dataset chloroset and try again and it works. So It seems that in case of bug just try the test dataset first.
Nice inpiration 2021_01| Comparative and phylogenetic analyses of the chloroplast genomes of species of Paeoniaceae Nice inpiration 2021_02 | Chloroplast genome evolution in the Dracunculus clade (Aroideae, Araceae)
Maximum Likelihood tree inference
Bayesian tree inference
For bayesian inference I found an online version of MrBayes tool. Amazing! So straithforward!
Extract tRNA and rRNA
To complete this task I saw a wonderfull tool available online and also in command line fashion.
Just upload select tRNA and get the fasta file. Very easy. Thanks to FeatureExtract developper.
But wait !!!! tRNA and rRNA are none-coding sequence. So you just need ATGC file! gbsextractor can do this task easily
Let's do it!
gbseqextractor \
-f Ceratotheca_sesamoides.gb \
-prefix cse -types tRNA
formating
cat cse.trna.fasta | awk '{gsub(/Ceratothec;/,"")}1' > cse.trna.formatted.fafind duplicated genes
awk '{if (x[$1]) { x_count[$1]++; print $0; if (x_count[$1] == 1) { print x[$1] } } x[$1] = $0}' cse.trna.formatted.fa | grep "^>" | sort
count the number
awk '{if (x[$1]) { x_count[$1]++; print $0; if (x_count[$1] == 1) { print x[$1] } } x[$1] = $0}' cse.trna.formatted.fa | grep "^>" | sort | wc -l
Explode
#[yedomon@localhost 04_sesamum_pedaloides]$ mkdir tRNA
#[yedomon@localhost 04_sesamum_pedaloides]$ cp cse.trna.formatted.fa tRNA/
#[yedomon@localhost 04_sesamum_pedaloides]$ cd tRNA/
cat cse.trna.formatted.fa | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fa")} print $0 > filename }'
mkdir doubling
mv trnA-UGC.fa trnI-CAU.fa trnI-GAU.fa trnL-CAA.fa trnN-GUU.fa trnR-ACG.fa trnV-GAC.fa doubling
cd doubling
for i in *.fa
do
base=$(basename $i .fa)
awk '{if(NR>2) {print $0}}' $i > ${base}.unik.fa
done
reformat
mkdir unik
cp *.unik.fa unik
cd unik
for f in *.unik.fa; do
mv -- "$f" "${f%.unik.fa}.fa"
done
Then
cp *.fa /home/yedomon/data/01_ka_ks/09_ceratotheca_sesamoides/tRNA
cd /home/yedomon/data/01_ka_ks/09_ceratotheca_sesamoides/tRNA
for i in *.fa
do
base=$(basename $i .fa)
cat $i | awk '{sub(/>.*/,">cse"); print}' > ${base}.formatted.fa
done
mkdir formatted
cp *.formatted.fa formatted/
cd formatted/
grep "^>" *.fa | wc -l
grep "^>" *.fa
Extract the CDS proteins gene
gbseqextractor \
-f wild_ghana_GeSeqJob-20210701-112928_wild_ghana_GenBank.gb \
-prefix gha -types CDS -cds_translation
Format
cat gha.cds_translation.fasta | awk '{gsub(/wild_ghana;/,"")}1' > gha.cds_translation_formatted.faafind duplicated genes
awk '{if (x[$1]) { x_count[$1]++; print $0; if (x_count[$1] == 1) { print x[$1] } } x[$1] = $0}' gha.cds_translation_formatted.faa | grep "^>" | sort
count the number
awk '{if (x[$1]) { x_count[$1]++; print $0; if (x_count[$1] == 1) { print x[$1] } } x[$1] = $0}' gha.cds_translation_formatted.faa | grep "^>" | sort | wc -l
Note : divided by 2 later of course
Explode
#[yedomon@localhost 04_sesamum_pedaloides]$ mkdir faa
#[yedomon@localhost 04_sesamum_pedaloides]$ cp ped.cds_translation_formatted.faa faa/
#[yedomon@localhost 04_sesamum_pedaloides]$ cd faa/
cat gha.cds_translation_formatted.faa | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".faa")} print $0 > filename }'vERIFICATION OF DOUBLING IN EACH GENE
Tips:
During the annotation process make sure you get the translation of rps12 gene. To get this use reference as third part or include multiple annotator together. Check the genebank file in notebook and Ctrl + H to see if rps12 was translated. Use gbsextractor to get CDS and use my awk script to explode then check some doubling manually.
fOR DOUDLING REMOVING DO
awk '{if(NR>2) {print $0}}' accD.faa # remove lines 1 and 2 of accD gene file
For looping
mkdir doubling
mv accD.faa atpF.faa clpP1.faa ndhB.faa ndhK.faa pafI.faa psbC.faa \
rpl2.faa rpl23.faa rpoC2.faa rps12.faa rps16.faa rps7.faa ycf1.faa ycf15.faa ycf2.faa doubling
cd doubling
for i in *.faa
do
base=$(basename $i .faa)
awk '{if(NR>2) {print $0}}' $i > ${base}.unik.faa
done
reformat
mkdir unik
cp *.unik.faa unik
cd unik
for f in *.unik.faa; do
mv -- "$f" "${f%.unik.faa}.faa"
done
Then
cp *.faa /home/yedomon/data/01_ka_ks/14_wild_ghana/faa
cd /home/yedomon/data/01_ka_ks/14_wild_ghana/faa
for i in *.faa
do
base=$(basename $i .faa)
cat $i | awk '{sub(/>.*/,">gha"); print}' > ${base}.formatted.faa
done
mkdir formatted
cp *.formatted.faa formatted/
cd formatted/
grep "^>" *.faa | wc -l
grep "^>" *.faa
I got this How do I make the vi editor display or hide line numbers? in order to see the line number for deletion .
and this How to remove lines with specific line number from text file with awk or sed in Linux/unix for removing quickly the doubling
grep "^>" *.faa
Remove manually dougling gene with vi then ...>
How to format a set of fatsa file by replace all character begining by ">" by ">a_single_name (species name). Usefull for tree contruction
The synthax is
cat test.faa | awk '{sub(/>.*/,">cse"); print}' > test.out.faa # remplacer tout ce qui vient en commencany > par >cseSince I have multiple fileS, I define the following code:
#!/bin/bash
set -e
for i in *.faa
do
base=$(basename $i .faa)
cat $i | awk '{sub(/>.*/,">cse"); print}' > ${base}.formatted.faa
done
Verification
grep "^>" *.faaOutput
accD.formatted.faa:>cse
atpA.formatted.faa:>cse
atpB.formatted.faa:>cse
Now I need to rename each file by including the species name in order to easely concatenate later for the tree construction
#!/bin/bash
set -e
for f in *.faa; do
mv -- "$f" "${f%.faa}.ctri.faa"
done
Work nicely
Note to avoid to do this just do
#!/bin/bash
set -e
for i in *.faa
do
base=$(basename $i .faa)
cat $i | awk '{sub(/>.*/,">cse"); print}' > formatted/${base}.formatted.cse.faa
done
tree based on ycf1 gene only
I face an issue need to transform ycf1.formatted.faa to ycf1.cse.formatted.faa
for f in *.formatted.faa; do
mv -- "$f" "${f%.formatted.faa}.gha.formatted.faa"
done
cp ycf1.gha.formatted.faa /home/yedomon/data/01_ka_ks/15_files_for_phyloThe path of each formatted gene
cd /home/yedomon/data/01_ka_ks/09_ceratotheca_sesamoides/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_1
cd /home/yedomon/data/01_ka_ks/10_ceratotheca_triloba/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_1
cd /home/yedomon/data/01_ka_ks/02_sesamum_alatum/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_1
cd /home/yedomon/data/01_ka_ks/03_sesamum_angolense/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_1
cd /home/yedomon/data/01_ka_ks/04_sesamum_pedaloides/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_1
cd /home/yedomon/data/01_ka_ks/05_sesamum_radiatum/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_1
cd /home/yedomon/data/01_ka_ks/01_sesamum_indicum_goenbaek/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_1
cd /home/yedomon/data/01_ka_ks/11_sesamum_indicum_yuzhi11/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_1
cd /home/yedomon/data/01_ka_ks/12_sesamum_indicum_ansanggae/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_1
prepare the file for tree set 1
cd /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_1
mkdir formatted
cat accD.* > formatted/accD.ready.faa
cat atpA.* > formatted/atpA.ready.faa
cat atpB.* > formatted/atpB.ready.faa
cat atpE.* > formatted/atpE.ready.faa
cat atpF.* > formatted/atpF.ready.faa
cat atpH.* > formatted/atpH.ready.faa
cat atpI.* > formatted/atpI.ready.faa
cat ccsA.* > formatted/ccsA.ready.faa
cat cemA.* > formatted/cemA.ready.faa
cat clpP1.* > formatted/clpP1.ready.faa
cat infA.* > formatted/infA.ready.faa
cat matK.* > formatted/matK.ready.faa
cat ndhA.* > formatted/ndhA.ready.faa
cat ndhB.* > formatted/ndhB.ready.faa
cat ndhC.* > formatted/ndhC.ready.faa
cat ndhD.* > formatted/ndhD.ready.faa
cat ndhE.* > formatted/ndhE.ready.faa
cat ndhF.* > formatted/ndhF.ready.faa
cat ndhG.* > formatted/ndhG.ready.faa
cat ndhH.* > formatted/ndhH.ready.faa
cat ndhI.* > formatted/ndhI.ready.faa
cat ndhJ.* > formatted/ndhJ.ready.faa
cat ndhK.* > formatted/ndhK.ready.faa
cat pafI.* > formatted/pafI.ready.faa
cat pafII.* > formatted/pafII.ready.faa
cat pbf1.* > formatted/pbf1.ready.faa
cat petA.* > formatted/petA.ready.faa
cat petB.* > formatted/petB.ready.faa
cat petD.* > formatted/petD.ready.faa
cat petG.* > formatted/petG.ready.faa
cat petL.* > formatted/petL.ready.faa
cat petN.* > formatted/petN.ready.faa
cat psaA.* > formatted/psaA.ready.faa
cat psaB.* > formatted/psaB.ready.faa
cat psaC.* > formatted/psaC.ready.faa
cat psaI.* > formatted/psaI.ready.faa
cat psaJ.* > formatted/psaJ.ready.faa
cat psbA.* > formatted/psbA.ready.faa
cat psbB.* > formatted/psbB.ready.faa
cat psbC.* > formatted/psbC.ready.faa
cat psbD.* > formatted/psbD.ready.faa
cat psbE.* > formatted/psbE.ready.faa
cat psbF.* > formatted/psbF.ready.faa
cat psbH.* > formatted/psbH.ready.faa
cat psbI.* > formatted/psbI.ready.faa
cat psbJ.* > formatted/psbJ.ready.faa
cat psbK.* > formatted/psbK.ready.faa
cat psbL.* > formatted/psbL.ready.faa
cat psbM.* > formatted/psbM.ready.faa
cat psbT.* > formatted/psbT.ready.faa
cat psbZ.* > formatted/psbZ.ready.faa
cat rbcL.* > formatted/rbcL.ready.faa
cat rpl14.* > formatted/rpl14.ready.faa
cat rpl16.* > formatted/rpl16.ready.faa
cat rpl20.* > formatted/rpl20.ready.faa
cat rpl22.* > formatted/rpl22.ready.faa
cat rpl23.* > formatted/rpl23.ready.faa
cat rpl2.* > formatted/rpl2.ready.faa
cat rpl32.* > formatted/rpl32.ready.faa
cat rpl33.* > formatted/rpl33.ready.faa
cat rpl36.* > formatted/rpl36.ready.faa
cat rpoA.* > formatted/rpoA.ready.faa
cat rpoB.* > formatted/rpoB.ready.faa
cat rpoC1.* > formatted/rpoC1.ready.faa
cat rpoC2.* > formatted/rpoC2.ready.faa
cat rps11.* > formatted/rps11.ready.faa
cat rps12.* > formatted/rps12.ready.faa
cat rps14.* > formatted/rps14.ready.faa
cat rps15.* > formatted/rps15.ready.faa
cat rps16.* > formatted/rps16.ready.faa
cat rps18.* > formatted/rps18.ready.faa
cat rps19.* > formatted/rps19.ready.faa
cat rps2.* > formatted/rps2.ready.faa
cat rps3.* > formatted/rps3.ready.faa
cat rps4.* > formatted/rps4.ready.faa
cat rps7.* > formatted/rps7.ready.faa
cat rps8.* > formatted/rps8.ready.faa
cat ycf15.* > formatted/ycf15.ready.faa
cat ycf1.* > formatted/ycf1.ready.faa
cat ycf2.* > formatted/ycf2.ready.faa
# Use this awk code to merge multiple sequence alignment into one
cat *.mafft.trimal | awk -v RS=">" -v FS="\n" -v OFS="\n" '{for(i=2; i<=NF; i++) {seq[$1] = seq[$1]$i}}; END {for(id in seq){print ">"id, seq[id]}}' > combined.fasta
Set 2 include this
cd /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
cd /home/yedomon/data/01_ka_ks/09_ceratotheca_sesamoides/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
cd /home/yedomon/data/01_ka_ks/10_ceratotheca_triloba/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
cd /home/yedomon/data/01_ka_ks/02_sesamum_alatum/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
cd /home/yedomon/data/01_ka_ks/03_sesamum_angolense/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
cd /home/yedomon/data/01_ka_ks/04_sesamum_pedaloides/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
cd /home/yedomon/data/01_ka_ks/05_sesamum_radiatum/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
cd /home/yedomon/data/01_ka_ks/01_sesamum_indicum_goenbaek/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
cd /home/yedomon/data/01_ka_ks/11_sesamum_indicum_yuzhi11/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
cd /home/yedomon/data/01_ka_ks/12_sesamum_indicum_ansanggae/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
cd /home/yedomon/data/01_ka_ks/13_sesamum_schinzianum/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
cd /home/yedomon/data/01_ka_ks/14_wild_ghana/faa/formatted/
cp *.faa /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
cd /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
ls *.faa | wc -lconcatenate genes for the tree construction
cd /home/yedomon/data/01_ka_ks/15_files_for_phylo/set_2
mkdir formatted
cat accD.* > formatted/accD.ready.faa
cat atpA.* > formatted/atpA.ready.faa
cat atpB.* > formatted/atpB.ready.faa
cat atpE.* > formatted/atpE.ready.faa
cat atpF.* > formatted/atpF.ready.faa
cat atpH.* > formatted/atpH.ready.faa
cat atpI.* > formatted/atpI.ready.faa
cat ccsA.* > formatted/ccsA.ready.faa
cat cemA.* > formatted/cemA.ready.faa
cat clpP1.* > formatted/clpP1.ready.faa
cat infA.* > formatted/infA.ready.faa
cat matK.* > formatted/matK.ready.faa
cat ndhA.* > formatted/ndhA.ready.faa
cat ndhB.* > formatted/ndhB.ready.faa
cat ndhC.* > formatted/ndhC.ready.faa
cat ndhD.* > formatted/ndhD.ready.faa
cat ndhE.* > formatted/ndhE.ready.faa
cat ndhF.* > formatted/ndhF.ready.faa
cat ndhG.* > formatted/ndhG.ready.faa
cat ndhH.* > formatted/ndhH.ready.faa
cat ndhI.* > formatted/ndhI.ready.faa
cat ndhJ.* > formatted/ndhJ.ready.faa
cat ndhK.* > formatted/ndhK.ready.faa
cat pafI.* > formatted/pafI.ready.faa
cat pafII.* > formatted/pafII.ready.faa
cat pbf1.* > formatted/pbf1.ready.faa
cat petA.* > formatted/petA.ready.faa
cat petB.* > formatted/petB.ready.faa
cat petD.* > formatted/petD.ready.faa
cat petG.* > formatted/petG.ready.faa
cat petL.* > formatted/petL.ready.faa
cat petN.* > formatted/petN.ready.faa
cat psaA.* > formatted/psaA.ready.faa
cat psaB.* > formatted/psaB.ready.faa
cat psaC.* > formatted/psaC.ready.faa
cat psaI.* > formatted/psaI.ready.faa
cat psaJ.* > formatted/psaJ.ready.faa
cat psbA.* > formatted/psbA.ready.faa
cat psbB.* > formatted/psbB.ready.faa
cat psbC.* > formatted/psbC.ready.faa
cat psbD.* > formatted/psbD.ready.faa
cat psbE.* > formatted/psbE.ready.faa
cat psbF.* > formatted/psbF.ready.faa
cat psbH.* > formatted/psbH.ready.faa
cat psbI.* > formatted/psbI.ready.faa
cat psbJ.* > formatted/psbJ.ready.faa
cat psbK.* > formatted/psbK.ready.faa
cat psbL.* > formatted/psbL.ready.faa
cat psbM.* > formatted/psbM.ready.faa
cat psbT.* > formatted/psbT.ready.faa
cat psbZ.* > formatted/psbZ.ready.faa
cat rbcL.* > formatted/rbcL.ready.faa
cat rpl14.* > formatted/rpl14.ready.faa
cat rpl16.* > formatted/rpl16.ready.faa
cat rpl20.* > formatted/rpl20.ready.faa
cat rpl22.* > formatted/rpl22.ready.faa
cat rpl23.* > formatted/rpl23.ready.faa
cat rpl2.* > formatted/rpl2.ready.faa
cat rpl32.* > formatted/rpl32.ready.faa
cat rpl33.* > formatted/rpl33.ready.faa
cat rpl36.* > formatted/rpl36.ready.faa
cat rpoA.* > formatted/rpoA.ready.faa
cat rpoB.* > formatted/rpoB.ready.faa
cat rpoC1.* > formatted/rpoC1.ready.faa
cat rpoC2.* > formatted/rpoC2.ready.faa
cat rps11.* > formatted/rps11.ready.faa
cat rps12.* > formatted/rps12.ready.faa
cat rps14.* > formatted/rps14.ready.faa
cat rps15.* > formatted/rps15.ready.faa
cat rps16.* > formatted/rps16.ready.faa
cat rps18.* > formatted/rps18.ready.faa
cat rps19.* > formatted/rps19.ready.faa
cat rps2.* > formatted/rps2.ready.faa
cat rps3.* > formatted/rps3.ready.faa
cat rps4.* > formatted/rps4.ready.faa
cat rps7.* > formatted/rps7.ready.faa
cat rps8.* > formatted/rps8.ready.faa
cat ycf15.* > formatted/ycf15.ready.faa
cat ycf1.* > formatted/ycf1.ready.faa
cat ycf2.* > formatted/ycf2.ready.faa
ycf1 gene DNA barcoding
- ycf1, the most promising plastid DNA barcode of land plants
- ycf1-ndhF genes, the most promising plastid genomic barcode, sheds light on phylogeny at low taxonomic levels in Prunus persica
- A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: a case study on ginsengs
- Chloroplast genome of white wild chrysanthemum, Dendranthema sp. K247003, as genetic barcode
/home/yedomon/utils/mafft-7.475-with-extensions/core/mafft all_ycf1.fna > all_ycf1.mafft
Ensuite je suis allez vers Bioedit pour visualiser where il y a des points communs.
Ensuite j'ai construit un fichier consensus issu de l'alignment avec emboss cons puis avec primer 3 online j'ai fait le design des marqueurs em mettant comme settings
targets : 4950,350. J'ai pu avoir un bon resultat en termes de la taille du marqueur (environ 500 bp). C'est deja cool je crois.
nucleotide diversity analysis
all codes and figure are here
mVista analysis
To plot mVista plot please do:
- Use the gff file
- filter by selecting gene, CDS, tRNA, strand, rRNA
- keep gene name intact by avoiding additionnal info like -2 or something else
- use filter fonction to make < and > symbole following strand + or -
- select a new column and use concatenate fonction to bring requeried field following
< 106481 116661 gene1
106481 106497 utr
107983 108069 exon
109884 110033 exon
111865 112023 exon
> 39424 42368 gene2
39424 39820 exon
41401 42368 exon
> 77817 81088 gene3
77817 78820 utr
79538 80107 exon
-
gene will be the name of the gene
-
utr will be tRNA and rRNA
-
exon will be CDS
-
then change gene name into utr and exon accordingly and remove < and > sign also just using filter function
that's it.
Tip: Later if you have n sequences to compare, enter n+1 and double the first one in order to get your four sequences fully.
IRSCOPE analysis
Some key points to successfully make Chloroplast junction graph with IRSCOPE R shiny tool
- Use the gff annotation from chloe or Geseq.
- Change the extension into .txt then copy and paste in Excel.
- Filter only by gene.
- Arrange the table as follow: start | end | gene name | strand.
- Delete IR annotation.
- Using chloe gff file is more flexible than geseq because of duplication of certain gene by Geseq tool
- save as tab delimitated file
- Make sure to arrange the fasta file as follow: LLS-IRb-SSC-IRb. Very important to run easily
- prepare the coordinates of junctions by doing real start of IRB-1basepair, real end of IRB, real start of IRA-1,real end of IRA
- Go to
https://irscope.shinyapps.io/irapp/then select upload data > Manual and upload the gff.txt, fasta and write down the coordinnates. Dependeing of the number of species, the process will take less than 10s.
Complete chloroplast genome of Myracrodruon urundeuva and its phylogenetics relationships in Anacardiaceae family
The strategies of plants against the adversities of environment may lead sequence evolution of cp genomes, with some genes under positive selection in numerous plant lineages (Rockenbach et al. 2016; Wu et al. 2020). The selective pressure can be measured as a ratio (ω) of the nonsynonymous substitution rate (dN) to the synonymous substitutions rate (dS) and using the site-model, we can assess the variability at different sites of a gene by comparing different models of evolution, such nearly neutral against selection by a LRT (Nielsen and Yang 1998; Jeffares et al. 2014; Wu et al. 2020). We did not detect evidence of branch-site selection, which reflects that M. urundeuva is not evolving faster than the other species in the family. On the other hand, significant evidence from site-model selection indicates that there are nine genes which are under positive selection, with the most genes under purifying selection in Anacardiaceae family. Several plastid genes were reported to be under selective pressure from different lineages of angiosperms, such clpP and accD (Erixon and Oxelman 2008; Rockenbach et al. 2016). In our study, we found that clpP gene has at least three codons under positive selection, but none related to accD gene. Furthermore, approximately 45% of genes from photosystem are under positive selection (ndhB, ndhD, rbcL and petD). In Chrysosplenium and Oryza genera, several genes related to photosynthesis (such rbcL and psbB) are associated to the environment due to high levels of UV radiation, which may lead to DNA damages and mutations, resulting in high mutation levels (Gao et al. 2019b; Wu et al. 2020). In fact, rbcL was proposed as a DNA barcoding marker in association with matK from Consortium for the Barcode of Life (CBOL) Plant Working Group (CBOL 2009). Dong et al. (2015) also reported high levels of discrimination success in plants based in rbcL and matK markers, and also proposed ycf1 as a new universal DNA barcoding marker, but in this work, the LRT revealed that ycf1 was not significant in the family. Thus, in addition to the genes already proposed as discriminating species in the literature, there is a need for intraspecific investigation of these positively selected genes identified in this work at the population level to assess their potential as markers for use in species identification
Ka ks analysis
Tutorial for branch model site
Prepare the datasets | nucleotide fasta and protein translated fasta files
Ceratotheca sesamoides
Extract the CDS proteins gene
gbseqextractor \
-f ansanggae_GeSeqJob-20210704-200810_Sesamum_indicum_cv_Ansanggae_GenBank.gb \
-prefix ansan -types CDS -cds_translation
Format
cat ansan.cds.fasta | awk '{gsub(/Sesamum_in;/,"")}1' > ansan.cds_formatted.fnaExplode
cat cse.cds_translation_formatted.faa | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".faa")} print $0 > filename }'Sesamum indicum faa and fna extraction
gbseqextractor \
-f Sesamum_indicum.gb \
-prefix ind -types CDS -cds_translation
formating the gene head names
cat ind.cds.fasta | awk '{gsub(/Sesamum_in;/,"")}1' > ind.cds.formatted.fasta
cat ind.cds_translation.fasta | awk '{gsub(/Sesamum_in;/,"")}1' > ind.cds_translation.formatted.fasta
Explode the sequences
cat ind.cds.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fna")} print $0 > filename }'
cat ind.cds_translation.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".faa")} print $0 > filename }'
Sesamum alatum faa and fna extraction
gbseqextractor
-f Sesamum_alatum.gb
-prefix ala -types CDS -cds_translation
cat ala.cds.fasta | awk '{gsub(/Sesamum_al;/,"")}1' > ala.cds.formatted.fasta
cat ala.cds_translation.fasta | awk '{gsub(/Sesamum_al;/,"")}1' > ala.cds_translation.formatted.fasta
cat ala.cds.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fna")} print $0 > filename }'
cat ala.cds_translation.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".faa")} print $0 > filename }'
Sesamum angolense faa and fna extraction
gbseqextractor
-f Sesamum_angolense.gb
-prefix ang -types CDS -cds_translation
cat ang.cds.fasta | awk '{gsub(/Sesamum_an;/,"")}1' > ang.cds.formatted.fasta
cat ang.cds_translation.fasta | awk '{gsub(/Sesamum_an;/,"")}1' > ang.cds_translation.formatted.fasta
cat ang.cds.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fna")} print $0 > filename }'
cat ang.cds_translation.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".faa")} print $0 > filename }'
Sesamum pedaloides faa and fna extraction
gbseqextractor
-f Sesamum_pedaloides.gb
-prefix ped -types CDS -cds_translation
cat ped.cds.fasta | awk '{gsub(/Sesamum_pe;/,"")}1' > ped.cds.formatted.fasta
cat ped.cds_translation.fasta | awk '{gsub(/Sesamum_pe;/,"")}1' > ped.cds_translation.formatted.fasta
cat ped.cds.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fna")} print $0 > filename }'
cat ped.cds_translation.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".faa")} print $0 > filename }'
Sesamum radiatum faa and fna extraction
gbseqextractor
-f Sesamum_radiatum.gb
-prefix rad -types CDS -cds_translation
cat rad.cds.fasta | awk '{gsub(/Sesamum_ra;/,"")}1' > ra.cds.formatted.fasta
cat rad.cds_translation.fasta | awk '{gsub(/Sesamum_ra;/,"")}1' > ra.cds_translation.formatted.fasta
cat ra.cds.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fna")} print $0 > filename }'
cat ra.cds_translation.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".faa")} print $0 > filename }'
Andrographis paniculata faa and fna extraction
gbseqextractor
-f Andrographis_paniculata.gb
-prefix and -types CDS -cds_translation
cat and.cds.fasta | awk '{gsub(/NC_022451.2;/,"")}1' > and.cds.formatted.fasta
cat and.cds_translation.fasta | awk '{gsub(/NC_022451.2;/,"")}1' > and.cds_translation.formatted.fasta
cat and.cds.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fna")} print $0 > filename }'
cat and.cds_translation.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".faa")} print $0 > filename }'
Aphelandra knappiae faa and fna extraction
gbseqextractor
-f Aphelandra_knappiae.gb
-prefix aph -types CDS -cds_translation
cat aph.cds.fasta | awk '{gsub(/NC_041424.1;/,"")}1' > aph.cds.formatted.fasta
cat aph.cds_translation.fasta | awk '{gsub(/NC_041424.1;/,"")}1' > aph.cds_translation.formatted.fasta
cat aph.cds.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fna")} print $0 > filename }'
cat aph.cds_translation.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".faa")} print $0 > filename }'
Vitis vinifera faa and fna extraction
gbseqextractor
-f Vitis_vinifera.gb
-prefix vit -types CDS -cds_translation
cat vit.cds.fasta | awk '{gsub(/NC_007957.1;/,"")}1' > vit.cds.formatted.fasta
cat vit.cds_translation.fasta | awk '{gsub(/NC_007957.1;/,"")}1' > vit.cds_translation.formatted.fasta
cat vit.cds.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fna")} print $0 > filename }'
cat vit.cds_translation.formatted.fasta | awk '{ if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".faa")} print $0 > filename }'
atpH gene case
/home/yedomon/utils/mafft-7.475-with-extensions/core/mafft atpH.faa > atpH.aln.mafft
/home/yedomon/utils/pal2nal.v14/pal2nal.pl atpH.aln.mafft atpH.fna -output paml -nogap > atpH.aln.mafft.pal2nal
/home/yedomon/utils/paml4.9j/bin/codeml dn_ds.ctl
python /home/yedomon/data/01_ka_ks/00_starting_block/dnds-master/parse_codeml_output.py dn_ds.mlc > results_dn_ds_atpH.txt
/home/yedomon/utils/paml4.9j/bin/codeml alternative_model.ctl
/home/yedomon/utils/paml4.9j/bin/codeml null_model.ctl
psbK case
/home/yedomon/utils/mafft-7.475-with-extensions/core/mafft psbK.faa > psbK.aln.mafft
/home/yedomon/utils/pal2nal.v14/pal2nal.pl psbK.aln.mafft psbK.fna -output paml -nogap > psbK.aln.mafft.pal2nal
/home/yedomon/utils/paml4.9j/bin/codeml dn_ds.ctl
python /home/yedomon/data/01_ka_ks/00_starting_block/dnds-master/parse_codeml_output.py dn_ds.mlc > results_dn_ds_psbK.txt
/home/yedomon/utils/paml4.9j/bin/codeml alternative_model.ctl
/home/yedomon/utils/paml4.9j/bin/codeml null_model.ctl
After calculating the LRT go here for p value calculation by pasting the LRT value and the df value also. Or here is better there is also the R comand. In fact the Tutorial from evo3d used right tail chi square
I test it
Chi-square = 6.024178, df = 1
Right-tail p-value is 0.01411
R command: *pchisq(6.024178, 1, lower.tail=FALSE)* or 1-pchisq(6.024178, 1)
pchisq(6.024178, 1, lower.tail=FALSE)
A total of 66 CDSs presented in all the analysed species, and were used for identification of positive selection using the branch-site model [32]. CDSs of each gene were aligned according to their amino acid sequences with MEGA7 [58]. The branch-site model in the program codeml of the PAML v4.9 package [60] was used to assess potential positive selection in Chrysosplenium that was set as the foreground branch. Selective pressure is measured by the ratio (ω) of the nonsynonymous substitution rate (dN) to the synonymous substitutions rate (dS). A neutral branch-site model (Model = 2, NSsites = 2, Fix = 1, and Fix ω = 1) and an alternative branch-site model (Model = 2, NSsites = 2, and Fix = 0) were applied separately. The right-tailed Chi-square test was used to compute p-values based on the difference of log-likelihood values between the two models with one degree of freedom. Moreover, BEB method [61] was implemented to calculate the posterior probabilities for amino acid sites that are potentially under positive selection. A gene with a p-value < 0.05 and ω > 1 was considered as a positively selected gene. An amino acid site with posterior probabilities > 0.95 was considered as positively selected.
Phylogenomic Analysis and Dynamic Evolution of Chloroplast Genomes in Salicaceae
To identify the microstructural mutations of Juglans, the five aligned sequences were further analyzed using DnaSP v5 (Librado and Rozas, 2009) and MEGA v5.0 (Tamura et al., 2011). Indel and SNP events were counted and positioned in the cp genome using DnaSP v5. Signatures of natural selection were studied for every chloroplast gene located outside of the inverted repeats region. Selective pressures (KA/KS) were computed with the codeml tool from PAML package v4.0 (Yang, 2007) using a YN00 model to test every gene sequence. We used the KaKs_calculator program to check the selective pressures (KA/KS) using same model as YN (Zhang et al., 2006). To avoid potential convergence biases, those genes with few mutations were filtered out from selective pressure analysis.
Chloroplast
Re-arrange multiple columns in a data set into one column using R
-
Comparative analysis of the complete chloroplast genome sequences of three Amaranthus species the PDF here
-
New | Completion of the Chloroplast Genomes of Five Chinese Juglans and Their Contribution to Chloroplast Phylogeny
The Juglans Cpg sequences from the finalized data set were aligned with MAFFT v7.0.0 (Katoh and Standley, 2013). The analyses were carried out based on the following three data sets: (1) the complete cp DNA sequences; (2) protein coding sequences; (3) the introns and spacers.
-
Bayesian analysis in R | Babette github | Babette demo | The error we make in Bayesian phylogenetics today
-
REF HERE
Just found today a call for submission from IJMS and a nice paper published
-
Comparative chloroplast genome analysis
Evolutionary dynamics of the chloroplast genome sequences of six Colobanthus species
1 Syntheny
Genome synteny analysis of the eight Colobanthus plastomes (six genomes reported in this paper, and C. quitensis and C. apetalus that were previously characterized and deposited in NCBI25,26) was performed with the use of MAUVE v.1.1.1
2 Ka and Ks
The evolutionary rate of the plastome genes identified in all Colobanthus species (C. acicularis, C. affinis, C. apetalus, C lycopodioides, C. nivicola, C. pulvinatus, C. quitensis and C. subulatus) was analyzed. A total of 78 genes were selected to estimate the ratio of non-synonymous (Ka) to synonymous (Ks) substitutions. Colobanthus quitensis was the reference species. These genes were extracted and aligned separately using MAFFT v7.310. The values of Ka and Ks in the shared genes were calculated in DnaSP v.6.10.04. Genes with non-applicable (NA) Ka/Ks ratios were changed to zero.
3 nucleotide diversity
The sequences were aligned in MAFFT v.7.31096 to perform sliding window analysis and evaluate nucleotide diversity (π) in cp genomes using DnaSP v.6.10.0497. The step size was set to 50 base pairs, and window length was set to 800 base pairs.
4 Potential RNA editing sites
Potential RNA editing sites in the protein-coding genes of chloroplast genomes were predicted using the Predictive RNA Editor for Plants (PREP) suite98. The cutoff value for the analyzed Colobanthus species was set at 0.8, and 34 out of the 35 reference genes in PREP were used. rpl23 was not included in the analysis because it was not identified within the chloroplast genomes of the studied Colobanthus species. Two previously sequenced cp genomes of C. quitensis and C. apetalus25,26 were also included in this analysis.
5 Phylogenetic analysis
The available (24) complete chloroplast genomes representing Caryophyllaceae lineages and the cp genome of Arabidopsis thaliana as an outgroup were downloaded from the NCBI database to investigate the phylogenetic relationships among the studied representatives of the genus Colobanthus and the genera in the family Caryophyllaceae. The cp genomes used in phylogenetic analyses are presented in Table 3. The sequences of 73 shared protein coding genes were extracted using custom R script, and they were aligned in MAFFT v.7.310. Finally, 73 concatenated protein-coding gene sequences where used for phylogeny reconstruction by Bayesian Inference (BI) and Maximum-Likelihood (ML) method. The best-fit model of sequence evolution was identified in MEGA v.799, and the GTR + G + I model was selected. The BI analysis was performed in MrBayes v.3.2.6100,101, and the ML analysis was conducted in PhyML v.3.0102. Parameter settings were previously described by Androsiuk et al.26.
6
Pi Figure 5
Partitioning the dataset is an important step in phylogenetic analyses, because this process is known to affect the accuracy of phylogenetic inference (Lanfear et al. 2012). Accordingly, we selected the best-fitting partitioning scheme by using the greedy search algorithm in PartitionFinder ver. 1.1.1 (Lanfear et al. 2012). The optimal partitioning scheme split the sequence alignment into six subsets (Table 2).
Buena vista con mVISTA For an upcoming publication, a doctoral student and I want to visualize the sequence variability among several plastid genomes via the tool mVISTA. This tool is often employed in plastid phylogenomic studies and generally simple to use. However, if a user wishes to input custom annotations, it can be quite tricky to generate the correct input for the software. Hence, I automated the generation of the input files as described below.
An effective visualization of plastid genomes in mVISTA requires the x input genomes the user wants to visualize („input genomes“ hereafter; e.g., x=2) as well as a reference genome. For simplicity, a user could employ x1 as the reference genome. The input genomes should be in FASTA format, the reference genome in GenBank format.
To generate custom annotations based on the reference genome, I employ the following Bash code, which (a) converts the reference genome from GenBank format to a cleaned GFF3 format (and incidentally also saves the genome in FASTA format), and (b) converts the cleaned GFF3 file to the mVISTA input.
NF=NC_000932.gb # Just an example
## Converting input file from GenBank format to a cleaned GFF3 format
# Note: This step also generates a FASTA file
grep -vE "codon_start|db_xref|exception" $INF > ${INF}2
to-gff --getfasta ${INF}2 ${INF%.gb*}.gff
grep "gene=" ${INF%.gb*}.gff | \
awk -F';' '{print $1}' | \
grep -vE "remark|intron|misc_feature|repeat_region" > ${INF%.gb*}.gff.clean
rm ${INF}2 ${INF%.gb*}.gff
## Converting input file from GFF3 format to VISTA format
grep -v "^#" ${INF%.gb*}.gff.clean | \
grep -v "rps12" | \
awk '{if ($3 ~ /gene/) {print $7" "$4" "$5" "$3" "$9} else {print $7" "$4" "$5" "$3} }' | \
awk '{if ($4 ~ /gene/) {gsub(/\+/, ">", $1); gsub(/\-/, "<", $1); print $0} else {$1=""; print $0} }' | \
sed 's/gene=//' | \
sed 's/gene/agene/' | \
sort -n -k2 | \
sed 's/agene/gene/' | \
awk '{$1=$1}1' | \
sed 's/CDS/exon/' | \
sed 's/tRNA/utr/' | \
sed 's/rRNA/utr/' > ${INF%.gb*}.mvista
-
My ultimate analyses reference from Bioinformaticians of Indian | Trichopus zeylanicus
-
The complete chloroplast genome of Gleditsia sinensis and Gleditsia japonica: genome organization, comparative analysis, and development of taxon specific DNA mini-barcodes
-
The Complete Chloroplast Genome Sequences of 14 Curcuma Species: Insights Into Genome Evolution and Phylogenetic Relationships Within Zingiberales
-
GOOD PLANT PLASTOME ANNOTATION TOOL | THESIS HERE
-
un bon argumentaire ..le graph des chloroplast la avec la perte du gene en question
-
Le graph de dn/ds et le choix de l'assemblage
-
Simple et clair a suivre
-
vraiment interessant paper chloroplast SNP origins geography paper
-
commentaire des resultats
-
Use of novoplasty for chloroplast assembly | Example 1 | Example 2 | Example 3 | Example 4 | Example 5 | Example 6 | Example 7 | Example 8 | Example 9 | Example 10
-
Distribution de mes samples
-
Sesamum indicum
-
Sesamum alatum
-
Sesamum angolense
-
Sesamum radiatum
-
Sesamum pedaloides | Page 25
-
Parfait exemple using chloroplast to solve taxonomic dispute | Idee Map pour ton article | Taxonomy and biogeography of Diapensia (Diapensiaceae) based on chloroplast genome data
-
2020 Bel argument en faveur du choix des CDs pour le phylogenetic analysis
Only the homologous CDs were used to construct the phylogenetic relationship to reduce data redundancy...
-
2020 Wahooo nice surprise | Only One species in Genomis Elselvier | Characterization of Withania somnifera chloroplast genome and its comparison with other selected species of Solanaceae
-
A second example from Genomics elselvier | Chloroplast genome sequences of Artemisia maritima and Artemisia absinthium: Comparative analyses, mutational hotspots in genus Artemisia and phylogeny in family Asteraceae
-
A third example: Evolutionary dynamics of chloroplast genomes in subfamily Aroideae (Araceae)
Chloroplast genomes, in circular form, were annotated using GeSeq [50] with BLAT [51] search of 85% identify for annotation of protein-coding genes, rRNAs and tRNAs. The annotations of tRNAs were further confirmed by using tRNAscan-SE v.2.0.3 [52] and ARAGORN v1.2.38 [53] by selection of default parameters of chloroplast genome. The position of start and stop codons were further confirmed by manual visualisation and Blast search with homologous genes of chloroplast genomes. Furthermore, the annotations of tRNAscan-SE v.2.0.3 [52] were preferred as compared to ARAGORN v1.2.38 [53] due to availability of its recently updated version. The five-column tab-delimited table of the annotation were generated for submission to National Center for Biotechnology Information (NCBI) through GB2sequin [54]. Fully annotated plastome circular diagrams were drawn by OrganellarGenomeDRAW (OGDRAW) [55]. These genomes were submitted to GenBank of National Center of Biotechnology Information (NCBI) under specific accession numbers (Table 1).
Find SNP from an alignment of two seqs
The solution is here
Assuming that you have your alignment in a file named "fasta.fas" this should get you started
from Bio import AlignIO
y=0
alignment = AlignIO.read("fasta.fas", "fasta")
for r in range(0,len(alignment[1].seq)):
if alignment[0,r] != alignment[1,r]:
if alignment[0,r] != "-" and alignment[1,r] != "-":
y=y+1
print r, alignment[0,r], alignment[1,r], y
else:
y=0
This returns position of SNP, nt in seq_A, nt in seq_B, running tally of the number of SNPs upstream of each indel
I tested this script in my server
First I align two chloroplast genome using mafft following this code
#---Convatenate both sequences
cat cp1.fasta cp2.fasta > cp1_cp2.fasta
#---Alignment
mafft --auto --thread 96 cp1_cp2.fata > cp1_cp2.mafft
#---Creating the python script
vi snp.py
#--Copy and past the python and run
python snp.py
I got this error
File "snp.py", line 8
(print r, alignment[0,r], alignment[1,r], y)
^
SyntaxError: invalid syntax
So I found a solution here
Then I modified the script just a little
from Bio import AlignIO
y=0
alignment = AlignIO.read("fasta.fas", "fasta")
for r in range(0,len(alignment[1].seq)):
if alignment[0,r] != alignment[1,r]:
if alignment[0,r] != "-" and alignment[1,r] != "-":
y=y+1
print (r, alignment[0,r], alignment[1,r], y)
else:
y=0
Note: The problem was that I should put in parenthesis all characters after print in line 8.
So I tried again
python snp.py
and I got
10 a t 1
11 a g 2
92 t c 3
113 a t 4
161 a c 1
234 c t 2
243 c t 1
246 a t 2
247 a t 3
248 a t 4
249 a t 5
266 t c 6
555 g a 7
1053 t c 8
1161 a g 9
1284 g a 10
1526 c t 11
1611 a c 12
1924 c g 1
1953 a t 1
1973 t g 2
2062 c g 3
2076 a g 4
2098 g t 5
2101 c g 6
2112 a g 7
2153 t g 8
2250 g t 9
2285 t g 10
2300 a g 11
2437 c g 12
2570 c a 13
2703 a c 14
2719 c t 15
2769 a c 16
2797 c t 17
2808 g c 18
3021 t c 19
3050 g c 20
3077 c t 21
3192 g a 22
3193 a c 23
3261 g c 24
3301 t g 25
3393 g a 26
3400 c g 27
3521 g a 28
3547 g t 29
3621 a g 30
3967 t g 1
4020 a t 2
4028 t g 3
4147 t c 4
4155 a c 5
4194 t c 6
4212 t g 7
4400 a c 8
4465 g t 9
4581 a c 1
4587 g a 2
4683 c t 3
4717 t g 1
4741 c a 1
4840 g a 2
4841 a g 3
4843 g a 4
4844 a g 5
4846 t a 6
4848 g t 7
4849 a g 8
4858 g t 9
4860 t g 10
4880 g a 11
4916 g t 12
4925 g t 13
4995 a g 14
5202 a g 1
5589 a g 2
5592 t c 3
5608 g c 4
5612 g a 5
5655 c t 6
5773 a t 7
5852 c a 1
5884 c g 2
5925 c a 3
5984 g a 4
6111 g c 5
6112 a g 6
6122 g a 7
6283 t a 8
6366 g t 9
6424 c a 1
6480 t g 2
6536 t c 3
6608 t a 1
6662 t g 2
6723 t c 3
6862 c t 1
6893 t a 1
6964 a t 2
7004 c t 3
7025 a c 4
7026 a c 5
7155 g c 6
7310 t c 7
7380 t c 8
7435 g a 9
7526 a c 10
7900 c t 11
7912 g t 12
8098 a t 13
8272 t g 14
8304 c g 15
8806 c a 1
9011 a t 2
9083 a c 1
9114 g t 2
9115 a g 3
9116 a t 4
9118 g a 5
9119 a c 6
9120 c t 7
9121 t a 8
9122 a g 9
9124 t c 10
9126 a t 11
9127 c t 12
9128 a c 13
9162 t c 14
9181 g a 15
9183 g a 16
9187 t c 17
9238 a c 18
9315 a c 19
9368 g t 20
9555 a t 21
9710 g a 22
9865 g c 23
9972 c t 1
10024 c g 2
10129 t c 3
10234 c a 4
10311 a c 5
10469 t a 1
10717 g a 2
10817 a c 3
10821 a g 4
10844 a g 5
11064 t g 6
11076 a g 7
11379 c g 8
11394 c a 9
11442 t c 10
11458 c g 11
11768 t c 12
11961 a g 13
12198 c t 14
12578 t c 15
12665 a c 16
13037 a g 1
13085 a c 2
13557 a c 1
13575 g t 2
13645 c t 3
13687 t g 4
13750 t c 5
13751 g a 6
14074 c g 7
14102 t c 8
14206 a c 9
14207 g a 10
14345 c g 11
14483 t g 12
14535 t c 13
14548 c t 14
14567 a c 15
14574 a c 16
14578 g a 17
14646 a t 18
14817 g a 19
14970 t a 1
15381 a g 1
15792 a c 1
15872 g t 2
15928 c t 3
15983 g t 1
16085 g a 2
16352 a g 3
16499 g a 4
16634 t c 5
16740 t c 6
16741 c a 7
16761 c a 8
16906 t c 9
17285 c g 1
17361 g a 2
18044 t a 3
18381 g a 4
18855 g c 5
18981 g c 6
18987 c t 7
18988 g t 8
19106 c t 9
19434 a g 10
19814 a g 11
19838 g t 12
19979 c g 13
20006 t g 14
20165 a c 15
20212 g t 16
20414 g a 17
20564 c a 18
20845 t c 19
21017 t a 20
21182 c t 21
21417 a c 22
21971 g c 23
22098 t g 24
22668 g t 25
22840 g a 26
23013 t c 27
23458 a c 28
23459 c a 29
23601 c t 30
24256 c t 31
24988 c t 32
25079 t c 33
25093 t c 34
25193 t c 35
25329 a g 36
25455 c t 37
25567 c t 38
25772 a g 39
25943 c t 40
26462 a g 41
26975 t a 42
27143 c a 43
27458 c t 44
27547 t c 45
27553 a g 46
27683 a c 47
27698 t g 48
27711 t g 49
27842 g a 1
27968 t c 2
28052 g a 3
28249 c t 4
28366 a g 5
28464 a g 6
28667 g c 1
28690 a g 2
28768 a g 3
28816 g a 1
28872 g c 2
28902 g t 3
28906 c t 4
28945 g a 5
28974 c t 1
29008 t c 2
29028 c t 3
29069 g a 4
29114 t c 5
29207 g c 6
29289 c t 7
29356 t c 8
29538 a g 9
29643 t g 10
29741 a c 1
29791 t c 2
29808 g c 1
29906 g c 2
29954 g a 3
29964 a t 4
30040 a c 5
30148 c a 6
30248 g a 7
30253 a c 8
30436 t g 9
30437 c a 10
30484 a g 11
30816 a c 1
30892 g a 2
30897 c a 3
30939 g c 4
31039 t c 5
31042 a g 1
31045 g a 2
31048 a g 3
31189 g t 4
31362 a c 5
31575 t g 6
31610 t g 7
31719 t g 8
31733 c a 9
32012 t c 1
32015 g t 2
32031 a t 3
32211 t c 1
32264 c g 2
32350 g a 1
32360 t g 2
32461 g a 3
32922 t c 4
32994 g a 5
33013 g t 6
33170 g a 7
33216 t g 8
33338 g a 9
33388 a g 10
33525 g a 11
34188 a c 12
34479 t c 13
34597 c t 14
34612 g a 15
34742 c t 16
35125 g a 17
35970 c a 18
36280 t c 1
36487 c g 1
36852 t c 1
36930 a t 1
36931 a c 2
37122 c a 3
37171 g a 1
Great! That works perfectly!
Furthermore, comparison of the cp genome of C. gileadensis with C. foliacea, C. wightii and B. sacra revealed 3,032, 8,787 and 5,120 SNPs as well as 3,580, 10,460 and 17,122 Indels respectively (Fig 4). Similarly, the C. foliacea cp genome also showed 8,194 and 5,182 SNPs while 7,632 and 17,970 Indel with C. wightii and B. sacra respectively. These Results shows that even the most conserved genome possesses some interspecific mutations which provides an important information in analyzing the phylogenetic and genetic diversity among the species
Indels detection
For indel detection I used the alignment file from the two species that I upload in DNAsp software. Then Ananlysis > Indels(Insertion-Deletion) polymorphism . YThen I got the following resut :
Input Data File: C:\...\indicum_triloba.mafft
Number of sequences: 2 Number of sequences used: 2
Selected region: 1-153439 Number of sites: 153439
Total number of sites (excluding sites with gaps / missing data): 152762
InDel Option: Model 1: Diallelic (non-overlapping)
Total number of InDel sites analysed: 677
Total number of (InDel and non-InDel) sites analysed, 152762 + 677: 153439
Total number of InDels events analysed, I: 90
Average InDel length event: 7.522
Average InDel length: 7.522
Number of InDel Haplotypes: 2
InDel Haplotype Diversity: 1.000
InDel Diversity, k(i): 90.000
InDel Diversity per site, Pi(i): 0.00059
Theta (per sequence) from I, Theta(i)-W: 90.000
The total number of indels sites here is 677. It is also possible to save the result in nexus file.
Similarly, a custom Python script (https://www.biostars.org/p/119214/) and DnaSP 5.10.01 [42], were employed to determine single-nucleotide polymorphisms and Indel polymorphisms among the complete genomes respectively.
To infer the phylogenetic position of both C. gileadensis and C. foliacea within the order Sapindales, 24 cp genomes were downloaded from the NCBI database for analysis. Multiple alignments were performed using complete cp genomes based on conserved structures and gene order [41] and 4 different methods were used to make the trees: Bayesian-inference (MrBayes v3.1.2 [43]), maximum parsimony (PAUP-4.0[44]), maximum-likelihood and neighbour joining (MEGA7.01[33]) according to the methods of Asaf et al [39,45]. For Bayesian posterior probabilities (PP) in the BI analyses, the best substitution model GTR + G model was tested according to the Akaike information criterion (AIC) by jModelTest verion 2102. The Markov Chain Monto Carlo (MCMC) was run for 1,000,000 generations with 4 incrementally heated chains, starting from random trees and sampling 1 out of every 100 generations. The first 30% of trees were discarded as burn-in to estimate the value of posterior probabilities. Furthermore, parameters for the ML analysis were optimized with a BIONJ tree as the starting tree with 1000 bootstrap replicates using the Kimura 2-parameter model with gamma-distributed rate heterogeneity and invariant sites. MP was run using a heuristic search with 1000 random addition sequence replicates with the tree-bisection-reconnection (TBR) branch-swapping tree search criterion. In the second phylogenetic analysis, 72 shared genes from the cp genomes of the twenty-six members of order Sapindales, were aligned using ClustalX with default settings, followed by manual adjustment to preserve reading frames. Similarly, the above4 mentioned phylogenetic inference models were utilized to build trees using 72 concatenated genes, using the same setting as described above and suggested by Asaf et al [45].
-
Very nice argument |
Previous studies based on plastid fragments and/or nuclear ribosomal DNA have had limited success resolving relationships within the genus Salvia. This study evaluates the efficacy of complete plastome sequences for phylogenetic inference within Salvia, using the recently established Salvia subg. Glutinaria as a case study. We use these plastomes to identify hypervariable and simple sequence repeat (SSR) regions for future studies within Salvia.The Chloroplast Genome of Salvia: Genomic Characterization and Phylogenetic Analysis -
2020 Only two | Very nice R graph Comparative analysis of chloroplast genomes in Vasconcellea pubescens A.DC. and Carica papaya L.
-
2020 Like my case Evolutionary and phylogenetic aspects of the chloroplast genome of Chaenomeles species
-
The Complete Chloroplast Genome Sequence of the Speirantha gardenii: Comparative and Adaptive Evolutionary Analysis
-
Species-tree inference with BEAST 2 and SNAPP
-
IRscope creator plastome paper
-
inverted repeat structural evolution in Rhus L
-
Simple tuto
-
Interessant title
-
Insights Into Chloroplast Genome Evolution Across Opuntioideae (Cactaceae) Reveals Robust Yet Sometimes Conflicting Phylogenetic Topologies
-
Best Practices/Softwares To Calculate Ka/Ks Ratio
-
The complete chloroplast genome of Stryphnodendron adstringens (Leguminosae - Caesalpinioideae): comparative analysis with related Mimosoid species
-
The Complete Chloroplast Genome Sequences of the Medicinal Plant Forsythia suspensa (Oleaceae)
-
Comparative Analysis of the Complete Chloroplast Genomes of Five Quercus Species
-
IR CONTRACTION
-
PSEUDOGENE
-
Inspiration
-
Dr swati 1
-
Dr swati 2
Nucleotide diversity work
I used DNAsp v6. First I aligned using MAFFT all fasta files then import the alignment file into DNAsp > analysis > DNA Polymorphism > click on compute > set window length to 800 bp and step size to 50 > Click OK.
Then move to R
I used the nucleotide diversity data and ran this code.
# Libraries
library(ggplot2)
# Data
data_pi = read.csv("pi.csv",sep = "," , h = T)
# Plot
p <- ggplot(data_pi, aes(x=Midpoint, y=Pi, colour = Pi)) +
ylim(0,0.033) +
geom_line() +
scale_colour_gradient2(low = "#0000b3", mid = "#3333ff" , high = "#ff0000", midpoint=median(data_pi$Pi)) +
#theme(legend.position="none") +
geom_hline(yintercept=0.017, color="#00b300", size=.4, linetype = "dotted" ) +
theme_minimal()
p
g <- ggplot(data_pi, aes(x=Midpoint, y=Pi, colour = Pi)) +
ylim(0,0.033) +
geom_line() +
scale_colour_gradient2(low = "#0000b3", mid = "#3333ff" , high = "#ff0000", midpoint=0.013) +
#theme(legend.position="none") +
geom_hline(yintercept=0.015, color="#00b300", size=.4, linetype = "dotted") +
labs(y = expression(Nucleotide~diversity~(pi)), x = "Midpoint position") +
theme_minimal()+
theme(panel.grid.major = element_line(colour = "grey", size = 0.2, linetype = "dotted")) +
theme(panel.grid.minor = element_line(colour = "grey", size = 0.2, linetype = "dotted")) +
theme(plot.background = element_rect(colour = "white", size = 1))+
theme(axis.text = element_text(colour = "black")) +
scale_x_continuous(limits=c(0,155000),
breaks = seq(0, 155000, by = 25000)) +
scale_y_continuous(limits=c(0,0.035),
breaks = seq(0, 0.035, by = 0.005))
and further process with Inkscape for arrow edition and annotation and get this. I used the g plot not the p.
ka ks work
cds extraction from genbank files of chloroplast data
Install gbseqextractor tool on centos 7
sudo yum search pip | grep python # to see availeble version
sudo yum install python3-pip.noarch
pip3 install gbseqextractor
Indicum
cd /home/kplee/analysis/0003_extraction/ind
gbseqextractor \
-f /home/kplee/datafiles/004_extraction/all_gb_files/Sesamum_indicum.gb \
-prefix indicum -seqPrefix ind -types CDS -cds_translation
### formating the gene head names
cat indicum.cds.fasta | awk '{gsub(/indSesamum_in;/,"ind_")}1' > ind.cds.formatted.fasta
### explode
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
alatum
cd /home/kplee/analysis/0003_extraction/ala
gbseqextractor \
-f /home/kplee/datafiles/004_extraction/all_gb_files/Sesamum_alatum.gb \
-prefix alatum -seqPrefix ala -types CDS -cds_translation
### formating the gene head names
cat alatum.cds.fasta | awk '{gsub(/alaSesamum_al;/,"ala_")}1' > ala.cds.formatted.fasta
### explode
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
angolense
cd /home/kplee/analysis/0003_extraction/ang
gbseqextractor \
-f /home/kplee/datafiles/004_extraction/all_gb_files/Sesamum_angolense.gb \
-prefix angolense -seqPrefix ang -types CDS -cds_translation
### formating the gene head names
cat angolense.cds.fasta | awk '{gsub(/angSesamum_an;/,"ang_")}1' > ang.cds.formatted.fasta
### explode
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
pedaloides
cd /home/kplee/analysis/0003_extraction/ped
gbseqextractor \
-f /home/kplee/datafiles/004_extraction/all_gb_files/Sesamum_pedaloides.gb \
-prefix pedaloides -seqPrefix ped -types CDS -cds_translation
### formating the gene head names
cat pedaloides.cds.fasta | awk '{gsub(/pedSesamum_pe;/,"ped_")}1' > ped.cds.formatted.fasta
### explode
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
radiatum
cd /home/kplee/analysis/0003_extraction/rad
gbseqextractor \
-f /home/kplee/datafiles/004_extraction/all_gb_files/Sesamum_radiatum.gb \
-prefix radiatum -seqPrefix rad -types CDS -cds_translation
### formating the gene head names
cat radiatum.cds.fasta | awk '{gsub(/radSesamum_ra;/,"rad_")}1' > rad.cds.formatted.fasta
### explode
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
now I will put indicum and alatum in a same directory for alignment
Hint: use a folder for ind and ala | put two by two by using cat ind_gene ala_gene > gene_name
then align using a loop for each gene
source activate mafft_env
mafft --maxiterate 1000 --localpair --thread 96 $genes > ${genes}.mafft
source deactivate mafft_env
Installation of PAML on Centos 7
$ wget http://abacus.gene.ucl.ac.uk/software/paml4.9j.tgz
$ tar xf paml4.9j.tgz
$ cd paml4.9j
$ rm bin/*.exe
$ cd src
$ make -f Makefile
$ ls -lF
$ rm *.o
$ mv baseml basemlg codeml pamp evolver yn00 chi2 ../bin
$ cd ..
Install PAL2NAL
wget http://www.bork.embl.de/pal2nal/distribution/pal2nal.v14.tar.gz
tar -xzvf pal2nal.v14.tar.gz
Install clustal-omaga
wget http://www.clustal.org/omega/clustalo-1.2.4-Ubuntu-x86_64
chmod u+x clustalo-1.2.4-Ubuntu-x86_64
newpub newpub2 newpub3 paper genome
Test ka ks calculation following this tutorial
git clone https://github.com/faylward/dnds
# Create an amino acid alignment
/home/kplee/program/clustalo/clustalo-1.2.4-Ubuntu-x86_64 -i cluster_1.faa -o cluster_1.aln.faa
# Convert aa alignment to na alignment
/home/kplee/program/pal2nal.v14/pal2nal.pl cluster_1.aln.faa cluster_1.fna -output paml -nogap > cluster_1.pal2nal
# Run codeml
# copy cluster_1.pal2nal and code.ctl into codeml executable file and run
codeml code.ctl
# Parse the output file
# I copied the output.txt into my dnds folder
python parse_codeml_output.py codeml.txt
work like a charm!
Now I will used my own data
#!/bin/bash
set -e
for i in *.faa
do
base=$(basename $i .faa)
/home/kplee/program/clustalo/clustalo-1.2.4-Ubuntu-x86_64 -i ${base}.faa -o ${base}.aln.faa
/home/kplee/program/pal2nal.v14/pal2nal.pl ${base}.aln.faa ${base}.fasta -output paml -nogap > ${base}.pal2nal
done
then mv all pal2nal file to the codeml path and set for gene one by one and get the results
work well
The synonymous (dS), non-synonymous (dN) nucleotide substitution rates and the dN/dS ratio (ω) were calculated using the codeml in Paml4.7 (Yang, 2007) with branch test model (Nielsen and Yang, 1998). Before analyses, the clade of Sino-Vietnamese species were set as foreground clade, and the others were set as background clade. The options of the two analyses were set to seqtype = 1, NSites = 0, model = 0 or model = 2. The likelihood ratio test (LRT) was used to estimate the quality of each model (Yang and Nielsen, 2002).
from here
Question: How to interpret LRT < 0 in codeml branch-model based test?
I am running codeml on orthogroup tree
The way I did is to set null model with fixed omega (model=2, NSsites=0, fix_omega=1) while branch-specific model by labeling branch of interest (foreground) in tree file (model=2, NSsites=0, fix_omega=0).
To found out the significant difference on Kn/Ks values between foreground and background branches, I calculate LRT by LRT=2(lnL1-lnL0)* and corresponding pvalues using dchisq function (df =1) in R.
Question: How to specify a branch in the tree automatically for branch-site model codeml?
4.5. Estimation of Evolutionary Rates The nucleotide sequences of 80 protein-coding genes were extracted from five Pogostemon cp genomes. Each gene was codon-aligned using the L-INS-I method in MAFFT v7. Phylogenetically informative characters (PICs) were counted for each gene using a Python script. Given both codon and related protein alignments of each gene, average nonsynonymous (dN) and synonymous (dS) substitution rates were estimated using the maximum likelihood method [55] with the F3 × 4 model implemented in the codeml program in PAML v4.9 [56]. In addition, protein-coding genes were assigned to nine functional groups according to the conventional classification. The genes within a functional group were concatenated for the above tests as well. The best ML tree based on PCGs was used as a constraint tree.
Definitively the most comprehensive tutorial about dn/ds logic and practical calculation
Theory
The selective pressure in protein coding genes can be detected within the framework of comparative genomics. The selective pressure is assumed to be defined by the ratio (ω) dN/dS. dS represents the synonymous rate (keeping the amino acid) and dN the non-synonymous rate (changing the amino acid). In the absence of evolutionary pressure, the synonymous rate and the non-synonymous rate are equal, so the dN/dS ratio is equal to 1. Under purifying selection, natural selection prevents the replacement of amino acids, so the dN will be lower than the dS, and dN/dS < 1. And under positive selection, the replacement rate of amino acid is favoured by selection, and dN/dS > 1.
Practical theory 1
CodeML and substitutions models: CodeML is a program from the package PAML, based on Maximum Likelihood, and developed in the lab of Ziheng Yang, University College London. It estimates various parameters (Ts/Tv, dN/dS, branch length) on the codon (nucleotide) alignment, based on a predefined topology (phylogenetic tree).
*My note: So a tree topology is a pre-requisite before selection pressure analysis. I need to know the tree topology of my taxa.
Practical theory 2
Different codon models exist in CodeML.
The model 0 estimates a unique dN/dS ratio for the whole alignment. Not really interesting, except to define a null hypothesis to test against.
Ah not really interesting ooohhhhhhhh!!!! Ah
The branch models estimate different dN/dS among lineages ie ASPM, a gene expressed in the brain of primates.
My case because I want to assess pressure selection among different taxa...So I think I will use thia option...
The site models estimate different dN/dS among sites (ie in the antigen-binding groove of the MHC).
The branch-site models estimate different dN/dS among sites and among branches. It can detect episodic evolution in protein sequences, as in the interactions between chains in the avian MHC. In my opinion, this is the most powerful application and this is the one used in the Sectome database (to which I contributed during my PhD).
What should I consider first ?
First, we have to define the branch where we think that position could have occurred **.
Note: In my case the foreground will be the branch were with my species of interest segregated. Like Dr Swati work. She wrote:
**The branch-site model was implemented by defining the Aster genus as foreground branch.**
the tree topology was this
We will call this branch the "foreground branch" and all other branches in the tree will be the "background" branches. The background branches share the same distribution of ω = dN/dS value among sites, whereas different values can apply to the foreground branch.
then
To compute the likelihood value, two models are computed: a null model, in which the foreground branch may have different proportions of sites under neutral selection to the background (i.e. relaxed purifying selection), and an alternative model, in which the foreground branch may have a proportion of sites under positive selection. As the alternative model is the general case, it is easier to present it first.
Theory about the brach-site model
Four categories of sites are assumed in the branch-site model:
Sites with identical dN/dS in both foreground and background branches:
K0 : Proportion of sites that are under purifying selection (ω0 < 1) on both foreground and background branches.
K1 : Proportion of sites that are under neutral evolution (ω1 = 1) on both foreground and background branches.
Sites with different dN/dS between foreground and background branches:
K2a: Proportion of sites that are under positive selection (ω2 ≥ 1) on the foreground branch and under purifying selection (ω0 < 1) on background branches.
K2b: Proportion of sites that are under positive selection (ω2 ≥ 1) on the foreground branch and under neutral evolution (ω1 = 1) on background branches.
For each category, we get the proportion of sites and the associated dN/dS values.
In the null model, the dN/dS (ω2) is fixed to 1:
Sites with identical dN/dS in both foreground and background branches:
K0 : Sites that are under purifying selection (ω0 < 1) on both foreground and background branches.
K1 : Sites that are under neutral evolution (ω1 = 1) on both foreground and background branches.
Sites with different dN/dS between foreground and background branches:
K2a: Sites that are under neutral evolution (ω2 = 1) on the foreground branch and under purifying selection (ω0 < 1) on background branches.
K2b: Sites that are under neutral evolution (ω2 = 1) on the foreground branch and under neutral evolution (ω1 = 1) on background branches.
Ah here a very important part after running codeml is to get Delta and the probability
For each model, we get the log likelihood value (lnL1 for the alternative and lnL0 for the null models), from which we compute the Likelihood Ratio Test (LRT). The 2×(lnL1-lnL0) follows a χ² curve with degree of freedom of 1, so we can get a p-value for this LRT.
The rest of the practical tuto is here
One example of result presentation is here. Anotherone is here
How to concatenate multiple sequence alignment file ? I was seeking an answr and I found An interesting tutorial on phylogenetic tree construction pipeline here. Definitively evomics is a very nice blog for genomics.
I found that phyutility is well used. How to download this java based tool? Here.
How to install? Just download it and uncompress the file. Since it is a java based tool, just run it by typing:
java -jar phyutility.jar -concat -in test.aln test2.aln -out testall.aln
an example from the tuto
phyutility -concat -in ~/workshop_materials/orthologs_concatenation/*.aligned.fa -out workshop_materials/orthologs_concatenation/concatenated_matrix.nexus
short explanation
Concatenating alignments together is a necessary task but can often be a painful assignment, especially when the same taxa are not found in all alignments. Phyutility will concatenate fasta or nexus files. Sequences will be concatenated as long as they have the same name between files. Each input file is considered a gene. Missing taxa in each gene will be given gaps for each gene in which the taxon is missing. The output is nexus or fasta and each needs to have been aligned separately before running.
So a hint: Make sure to name ideally the gene uniformely to facilitate the data matrix constitution and align eah genes orthologues separately before
pHYLOGENETIC CONSTRUCTION
Prepare the files
I used gbseqextractor tool
[kplee@localhost ~]$ gbseqextractor --help
usage: gbseqextractor [-h] -f <STR> -prefix <STR> [-seqPrefix <STR>]
[-types {CDS,rRNA,tRNA,wholeseq,gene} [{CDS,rRNA,tRNA,wholeseq,gene} ...]]
[-cds_translation] [-gi] [-p] [-t] [-s] [-l] [-rv] [-F]
Extract any CDS or rNRA or tRNA DNA sequences of genes from Genbank file.
Seqid will be the value of '/gene=' or '/product=', if they both were not
present, the gene will not be output!
version 20201128:
Now we can handle compounlocation (feature location with "join")!
We can also output the translation for each CDS (retrived from '/translation=')
Please cite:
Guanliang Meng, Yiyuan Li, Chentao Yang, Shanlin Liu,
MitoZ: a toolkit for animal mitochondrial genome assembly, annotation
and visualization, Nucleic Acids Research, https://doi.org/10.1093/nar/gkz173
optional arguments:
-h, --help show this help message and exit
-f <STR> Genbank file
-prefix <STR> prefix of output file. required.
-seqPrefix <STR> prefix of each seq id. default: None
-types {CDS,rRNA,tRNA,wholeseq,gene} [{CDS,rRNA,tRNA,wholeseq,gene} ...]
what kind of genes you want to extract? wholeseq for
whole fasta seq. WARNING: Each sequence in the result
files corresponds to ONE feature in the GenBank file,
I will NOT combine multiple CDS of the same gene into
ONE! [CDS]
-cds_translation Also output translated CDS (required -types CDS). The
translations are retrived directly from the
'/translation=' key word. [False]
-gi use gi number as sequence ID instead of accession
number when " gi number is present. (default:
accession number)
-p output the position information on the ID line.
Warning: the position on ID line is 0 left-most!
[False]
-t output the taxonomy lineage on ID line [False]
-s output the species name on the ID line [False]
-l output the seq length on the ID line [False]
-rv reverse and complement the sequences if the gene is on
minus strand. Always True!
-F only output full length genes,i.e., exclude the genes
with '>' or '<' in their location [False]
For Adenocalymma_acutissimum.gb
#--Extraction of CDS
gbseqextractor \
-f Adenocalymma_acutissimum.gb \
-prefix Adenocalymma -seqPrefix ade -types CDS -cds_translation
#--Format the header in species_gene style.
cat Adenocalymma.cds_translation.fasta | awk '{gsub(/NC_037455.1;/,"_")}1' > Adenocalymma.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Amphilophium_carolinae.gb
#--Extraction of CDS
gbseqextractor \
-f Amphilophium_carolinae.gb \
-prefix Amphilophium -seqPrefix amp -types CDS -cds_translation
#--Format the header in species_gene style.
cat Amphilophium.cds_translation.fasta | awk '{gsub(/NC_042933.1;/,"_")}1' > Amphilophium.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Anemopaegma_acutifolium.gb
#--Extraction of CDS
gbseqextractor \
-f Anemopaegma_acutifolium.gb \
-prefix Anemopaegma -seqPrefix ane -types CDS -cds_translation
#--Format the header in species_gene style.
cat Anemopaegma.cds_translation.fasta | awk '{gsub(/NC_037226.1;/,"_")}1' > Anemopaegma.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Fraxinus_excelsior.gb
#--Extraction of CDS
gbseqextractor \
-f Fraxinus_excelsior.gb \
-prefix Fraxinus -seqPrefix fra -types CDS -cds_translation
#--Format the header in species_gene style.
cat Fraxinus.cds_translation.fasta | awk '{gsub(/NC_037446.1;/,"_")}1' > Fraxinus.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Mentha_longifolia.gb
#--Extraction of CDS
gbseqextractor \
-f Mentha_longifolia.gb \
-prefix Mentha -seqPrefix men -types CDS -cds_translation
#--Format the header in species_gene style.
cat Mentha.cds_translation.fasta | awk '{gsub(/NC_032054.1;/,"_")}1' > Mentha.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Oleae_europaea.gb
#--Extraction of CDS
gbseqextractor \
-f Oleae_europaea.gb \
-prefix Oleae -seqPrefix ole -types CDS -cds_translation
#--Format the header in species_gene style.
cat Oleae.cds_translation.fasta | awk '{gsub(/NC_013707.2;/,"_")}1' > Oleae.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Pedicularis_longiflora.gb
#--Extraction of CDS
gbseqextractor \
-f Pedicularis_longiflora.gb \
-prefix Pedicularis -seqPrefix plo -types CDS -cds_translation
#--Format the header in species_gene style.
cat Pedicularis.cds_translation.fasta | awk '{gsub(/NC_046852.1;/,"_")}1' > Pedicularis.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Perilla_citriodora.gb
#--Extraction of CDS
gbseqextractor \
-f Perilla_citriodora.gb \
-prefix Perilla -seqPrefix pci -types CDS -cds_translation
#--Format the header in species_gene style.
cat Perilla.cds_translation.fasta | awk '{gsub(/NC_030755.1;/,"_")}1' > Perilla.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Salvia_splendens.gb
#--Extraction of CDS
gbseqextractor \
-f Salvia_splendens.gb \
-prefix Salvia -seqPrefix sal -types CDS -cds_translation
#--Format the header in species_gene style.
cat Salvia.cds_translation.fasta | awk '{gsub(/NC_050901.1;/,"_")}1' > Salvia.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Vitis_vinifera.gb
#--Extraction of CDS
gbseqextractor \
-f Vitis_vinifera.gb \
-prefix Vitis -seqPrefix vit -types CDS -cds_translation
#--Format the header in species_gene style.
cat Vitis.cds_translation.fasta | awk '{gsub(/NC_007957.1;/,"_")}1' > Vitis.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Ansanggae.gb
#--Extraction of CDS
gbseqextractor \
-f Sesamum_indicum_cv_Ansanggae.gb \
-prefix Ansanggae -seqPrefix ans -types CDS -cds_translation
#--Format the header in species_gene style.
cat Ansanggae.cds_translation.fasta | awk '{gsub(/NC_016433.2;/,"_")}1' > Ansanggae.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Goenbaek.gb
#--Extraction of CDS
gbseqextractor \
-f Goenbaek.gb \
-prefix Goenbaek -seqPrefix goe -types CDS -cds_translation
#--Format the header in species_gene style.
cat Goenbaek.cds_translation.fasta | awk '{gsub(/Sesamum_in;/,"_")}1' > Goenbaek.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Alatum.gb
#--Extraction of CDS
gbseqextractor \
-f alatum.gb \
-prefix Alatum -seqPrefix ala -types CDS -cds_translation
#--Format the header in species_gene style.
cat Alatum.cds_translation.fasta | awk '{gsub(/Sesamum_al;/,"_")}1' > Alatum.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Angolense.gb
#--Extraction of CDS
gbseqextractor \
-f Angolense.gb \
-prefix Angolense -seqPrefix ang -types CDS -cds_translation
#--Format the header in species_gene style.
cat Angolense.cds_translation.fasta | awk '{gsub(/Sesamum_an;/,"_")}1' > Angolense.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Pedaloides.gb
#--Extraction of CDS
gbseqextractor \
-f Pedaloides.gb \
-prefix Pedaloides -seqPrefix ped -types CDS -cds_translation
#--Format the header in species_gene style.
cat Pedaloides.cds_translation.fasta | awk '{gsub(/Sesamum_pe;/,"_")}1' > Pedaloides.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Radiatum.gb
#--Extraction of CDS
gbseqextractor \
-f Radiatum.gb \
-prefix Radiatum -seqPrefix rad -types CDS -cds_translation
#--Format the header in species_gene style.
cat Radiatum.cds_translation.fasta | awk '{gsub(/Sesamum_ra;/,"_")}1' > Radiatum.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Avicennia_marina.gb
#--Extraction of CDS
gbseqextractor \
-f Avicennia_marina.gb \
-prefix Avicennia -seqPrefix avi -types CDS -cds_translation
#--Format the header in species_gene style.
cat Avicennia.cds_translation.fasta | awk '{gsub(/NC_047414.1;/,"_")}1' > Avicennia.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Andrographis_paniculata.gb
#--Extraction of CDS
gbseqextractor \
-f Andrographis_paniculata.gb \
-prefix Andrographis -seqPrefix and -types CDS -cds_translation
#--Format the header in species_gene style.
cat Andrographis.cds_translation.fasta | awk '{gsub(/NC_022451.2;/,"_")}1' > Andrographis.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
For Aphelandra knappiae
#--Extraction of CDS
gbseqextractor \
-f Aphelandra_knappiae.gb \
-prefix Aphelandra -seqPrefix aph -types CDS -cds_translation
#--Format the header in species_gene style.
cat Aphelandra.cds_translation.fasta | awk '{gsub(/NC_041424.1;/,"_")}1' > Aphelandra.cds_translation_formatted.fasta
#--Explode and renamed with the corresponding header
cat * | awk '{
if (substr($0, 1, 1)==">") {filename=(substr($0,2) ".fasta")}
print $0 > filename }'
Get the common genes using Bioinformatics & Evolutionary Genomics venn diagram web tool
Got a total of 79 shared genes
atpH
psbK
ndhI
rpl20
matK
ndhF
atpF
psbF
petN
rps7
psbE
petB
rpl16
ycf2
petG
petA
rps12
rpl2
ndhH
clpP
rps3
rps2
atpB
ndhE
psaA
psbT
psbM
ycf1
infA
rpoB
rpl36
psbC
ndhD
rpoC1
atpE
accD
psbN
rps11
rps4
psaI
rps18
ndhC
psaJ
psbL
rpl33
ndhA
petL
rpl23
rps19
ndhG
ndhK
rpl32
rpl14
psaC
psbJ
rpl22
rps8
ndhB
psbH
rpoC2
ndhJ
rbcL
atpI
psbB
rpoA
ycf3
psbD
rps16
cemA
ccsA
psbZ
rps14
petD
atpA
rps15
psaB
psbA
psbI
ycf4
ycf15 is not present in Vitis vinifera from a set of: Rad,Ind,Ala,Ped,Ang,Vit,And,Aph.
Note: Need to change clP1 (from Geseq) ---> to clP and pbf1 (from Geseq) ---> to psbN
evomics workshop on multiple sequence alignement, concatenation , parttion and model testing and phylogenetic tree visualization
Amazing tutorials of Michael Matschiner for Phylogenetic inference | Michael Matschiner Publication