importosimportmatplotlib.pyplotaspltimportpandasaspdimportscanpyasscimportVDJclusteringasvdjc# Merge gex and vdj from multiple samples# 1st parameter: sample configuration tsvfile. The first 4 columns are mandatory: sample, gex_data, gex_data_type and vdj_data. The following columns are user optional and they are used to specify sample categories.# 2nd paramter: output folder. It must be empty.sample_config="input.tsv"out_path="./"adata=vdjc.read_gex_vdj(sample_config, out_path)
# We could also start with previous results# adata = sc.read_h5ad(os.path.join(out_path, "merged_gex_vdj.h5ad"))# Show cell numbers in different categoriesvdjc.pp.show_sample_category(adata, "input_short.tsv")
# If you want to select tumor samples, thenadata=adata[adata.obs["tissue"] =="tumor"]
adata.obs# Quality checkadata=vdjc.pp.gex_calculate_qc_metrics(adata, "mouse")
# filter out cells that have less than 200 genes expressed# filter out genes that are detected in less than 3 cells# filter out cells with percent mito >= 0.2# filter out cells with percent ribo <= 0.05adata=vdjc.pp.gex_filter(adata, min_genes=200, min_cells=3, 20, 5)
# normalize to depth 10 000# logaritmizeadata=vdjc.pp.gex_norm_logtrans(adata, counts_per_cell_after=1e4)
# Detect highly variable genesadata=vdjc.pp.highly_variable_genes_all(adata)
adata=vdjc.pp.highly_variable_genes_sample(adata)
# User could specify the threshold of selecting highly variable genes# For example, select highly variable genes that are common in at least half of the samplesthreshold=len(set(adata.obs['sample']))/2var_select=adata.var.highly_variable_nbatches>=thresholdvar_genes=var_select.index[var_select]
print(f"Detect {len(var_genes)} highly variable genes")
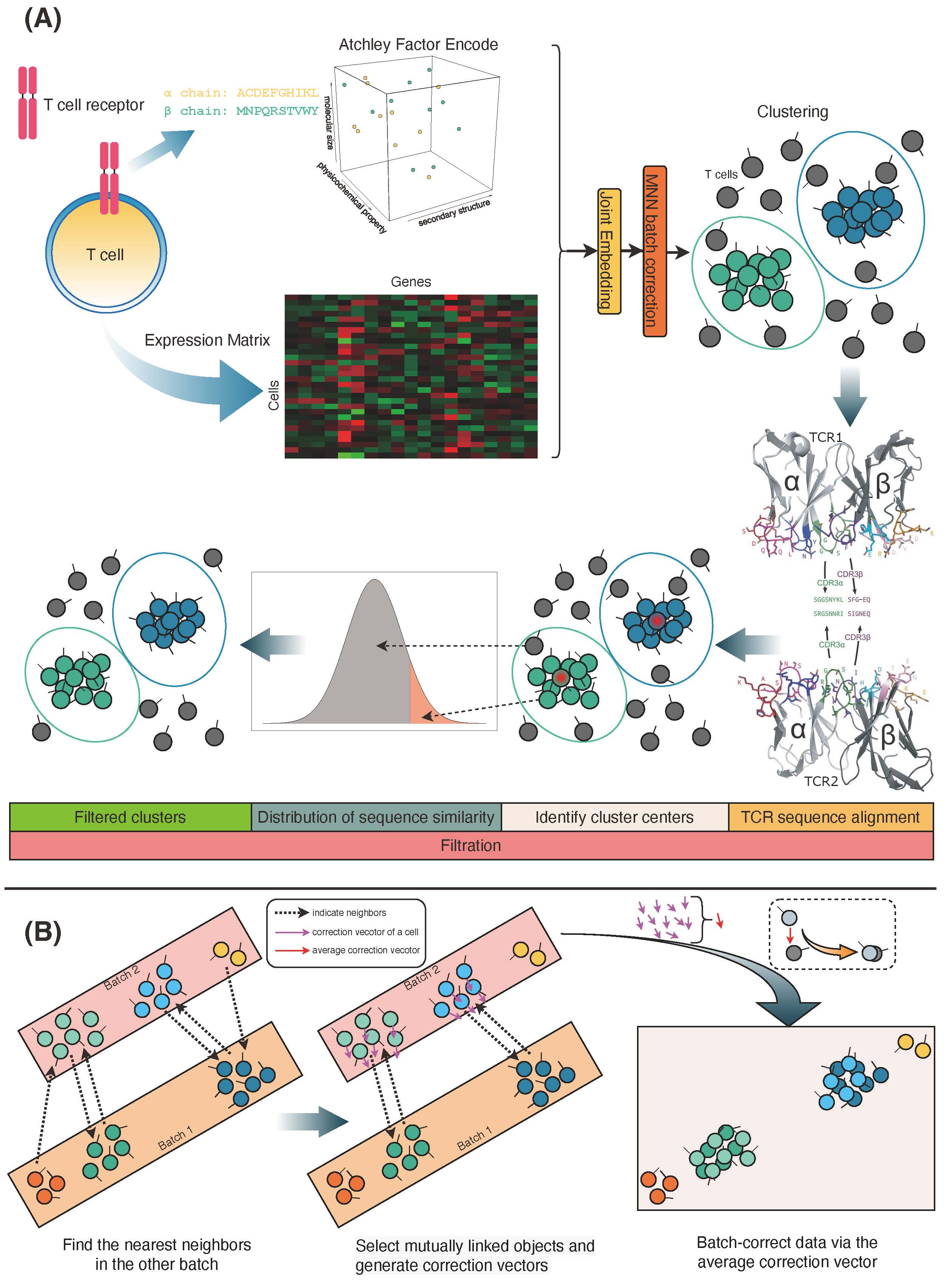
# Integrate GEX (count) and VDJ (Acthley factor)adata_gex_vdj=vdjc.pp.generate_gex_vdj(adata, var_genes)
# Bach correct using MNNcorr_data=vdjc.pp.batch_correct_mnn(adata_gex_vdj, sample_config)
# Z-score transformation: give each feature (GEX and VDJ) a similar weight corr_data_scaled=vdjc.pp.zscore_transform(corr_data)
# Calculate PCA and neighborhood graphcorr_data_scaled=vdjc.tl.pca_neighbors(corr_data_scaled)
# Plot UMAPvdj.pl.umap(corr_data_scaled, color="sample", title="MNN Corrected umap")
# Clusteringvdj.tl.leiden(corr_data_scaled, resolution=0.4, key_added="leiden_0.4")
# Filter cells in clusters# In order to speed up the process, you could skip the alignment step and recomputation of Atchley Factors in function vdjc.tl.centers_corrs. Therefore, you could use the other function vdjc.tl.centers_corrs_noalign.cen, cor=vdjc.tl.centers_corrs(corr_data_scaled, "leiden_0.4")
corr_data_scaled_filt=vdjc.tl.filter(corr_data_scaled, key="leiden_0.4", thr=0.9, centers=cen, corrs=cor)
# Filter clusterscen, cor=vdjc.tl.centers_corrs(corr_data_scaled_filt, "leiden_0.4")
corr_data_scaled_filt=vdjc.tl.filter(corr_data_scaled_filt, key="leiden_0.4", thr=0.9, centers=cen, corrs=cor)