Miekki
Minhash Index Extended to Knead Kmer Intersection

Description (bird's-eye view) :
Miekki is an index structure intended to index huge genome collections with reasonable resources. Given a query sequence, it can return the list of genomes that share more than S k-mers with it. The query sequence may be a gene, a transcript, a genome or a long read under certain conditions. The index constructs and stores a partition of fingerprints from each genome. The query sequence is also decomposed in a partition of fingerprints and compared to the relevant partition of the index. From the amount of shared fingerprints between each genome and the query, the k-mer intersection is ESTIMATED and returned if it is above a given threshold. The amount of fingerprints to store by genomes is the key parameter. More fingerprints will allow a better estimation of the intersection, but will also raise the index sensibility (it will be able to find matches from a lower amount of shared k-mers). Of course, more fingerprints also means a larger index and potentially a longer query time.
Usage:
The first try:
./miekki -l genome_list.txt -a query_sequence.fasta -o output.txt -d my_index_saved_for_later.gz
The second try that loads the index from disk:
./miekki -i my_index_saved_for_later.gz -a query_sequence.fasta -o output.txt
The real try with multiple cores (8) and a custom k-mer size (21):
./miekki -l genome_list.txt -a query_sequence.fasta -o output.txt -d my_index_saved_for_later.gz -t 8 -k 21
I do not find what all my matches, let index more fingerprints!
./miekki -l genome_list.txt -a query_sequence.fasta -o output.txt -d my_index_saved_for_later.gz -h 20
I have too many hits!!! I want matches from 10,000 shared k-mers:
./miekki -l genome_list.txt -a query_sequence.fasta -o output.txt -d my_index_saved_for_later.gz -s 10000
Help:
./miekki
This is a help message
Input
-i load a constructed index from disk
-l construct an index from a list of file
-a query a fasta file
Output
-o output file name (out.txt)
-d dump the index on disk
Performances
-h use 2^h minimizers per sequence (20 for 1048576 minimizers)
-k kmer size (31)
-s minimal estimated intersection to be reported (100)
-t thread number (1)
Advanced usage
-f fingerprint size
-b 2^b bits used for the bloom filter
-e exact mode, the real intersection will be computed on hits
Output
For each query sequence, Miekki will return a list of genomes (probably) sharing more than S kmers along with the fingerprint hit found, the approximated k-mer intersection and the Jaccard distance between the genome and the query.
The output is formated with one sequence per line starting by its header, followed by a list of genome with the associated metrics.
Header_query_sequence:genome_file_name1 shared_fingerprint shared_kmer_estimation jaccard_distance_estimation;genome_file_name2 shared_fingerprint shared_kmer_estimation jaccard_distance_estimation;\n
Sample Output
>read1:GCA_000007545.1_ASM754v1_genomic.fna.gz 7274 36544 0.007626;GCA_000007145.1_ASM714v1_genomic.fna.gz 390 1925 0.000409;GCA_000007645.1_ASM764v1_genomic.fna.gz 367 1558 0.000388;GCA_000007725.1_ASM772v1_genomic.fna.gz 365 1151 0.000401;GCA_000007605.1_ASM760v1_genomic.fna.gz 357 929 0.000412;
>read2:GCA_000007545.1_ASM754v1_genomic.fna.gz 221 783 0.000238;GCA_000007145.1_ASM714v1_genomic.fna.gz 173 493 0.000196;GCA_000007645.1_ASM764v1_genomic.fna.gz 167 709 0.000177;GCA_000007725.1_ASM772v1_genomic.fna.gz 159 482 0.000177;GCA_000007605.1_ASM760v1_genomic.fna.gz 154 391 0.000182;
>read3:GCA_000007545.1_ASM754v1_genomic.fna.gz 7465 37503 0.007826;GCA_000007145.1_ASM714v1_genomic.fna.gz 354 996 0.000402;GCA_000007645.1_ASM764v1_genomic.fna.gz 351 1106 0.000385;GCA_000007725.1_ASM772v1_genomic.fna.gz 341 861 0.000399;GCA_000007605.1_ASM760v1_genomic.fna.gz 332 1410 0.000351;
Fingerprint number
-h 17 will create 2^17=131,072 fingerprint per file.
(-h 18 262,144 -h 19 524,288 -h 20 1,048,576 and so on)
The index uses 1 byte to store a fingerprint (before compression) so -h 20 needs at most 1MB per file, -h19 two times less, etc...
To have a rough intuition of the index sensibility, 1M fingerprint means on average 1 fingerprint for 10 k-mers for a 10 megabase genome. We expect an errorless 1 kilobase query to have roughly a hundred fingerprints, and it is therefore very likely to be found by the index. With -h 17 we expect on average 1 fingerprint for 100 k-mers, so a 1 kilobase query may sometime be missed by the index.
As expected the more k-mers a query shares with a genome, the easier the match could be found. Therefore, larger queries tend to be found even if the index is very small. However, smaller queries could need larger indexes to be reliably found.
As a consequence query shorter than 1 kilobase may not be reliably found and the performances of Miekki on such sequences havee yet to be evaluated.
Exact mode
If you keep the indexed genomes on disk, you can actually perform exact queries with the -e option. The index willfirst compute the candidate genomes and therefore load them in memory to compute the exact intersection and jaccard. The output is formated as:
real_jaccard jaccard_estimation real_intersection_size intersection_size_estimation sequence_header genome_file_name
The correlation between the estimation and the real value can be plot with the R script ScatterMiekki.r to get plot like this:

Sample Output
0.0131596 0.0131626 37300 37820.5 >read1 GCA_000007625.1_ASM762v1_genomic.fna.gz
0.000150082 0.000245384 369 618.65 >read2 GCA_000007245.1_ASM724v1_genomic.fna.gz
Warning: This tools is a work in progress
Known problems/incoming features:
Union estimation is not accurate yet.
All genomes have the same amount of minimizers disregarding their sizes. It could be a problem when indexing genomes with unequal sizes. Two solutions: Split large genomes or use a COBS-like approach.
Column could be way more compressed. Other compression scheme could be tested. A clever ordering of the lines could allow a very efficient column compression and therefore a reduced memory usage AND a faster query.
Index construction is not efficiently parallel. An efficient method woudl be to construct several index in parallel and to concatenate them afterward.
Query time is not yet optimized. Batch queries could lead to a higher throughput.
Description (worm's-eye view)
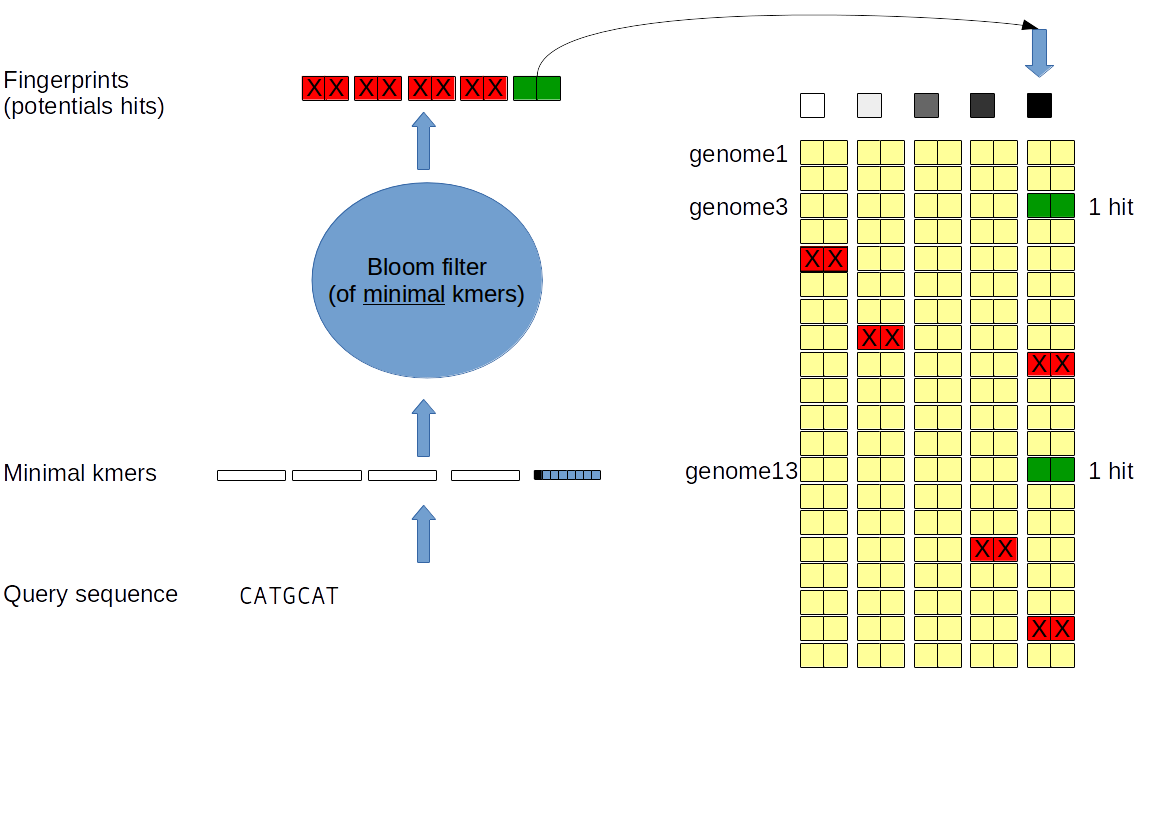
The index is composed of 2^h vectors of (hyperminhash) fingerprints and a (shifted) Bloom filter.
 Each vector is responsible of the (hyperminhash) fingerprints of a given partition.
Each of these vector have the same size that is the number of genome indexed.
The index can be seen as a matrix with column are partitions and line are genomes.
The Bloom filter contain all the kmers that have been inserted in the index.
Each vector is responsible of the (hyperminhash) fingerprints of a given partition.
Each of these vector have the same size that is the number of genome indexed.
The index can be seen as a matrix with column are partitions and line are genomes.
The Bloom filter contain all the kmers that have been inserted in the index.

For each genome, all kmers are hashed and partitionned according to the h first bit of their hashes. For each partition the minimal (hashed) kmer is inserted into a (shifted) bloom filter and its hyperminhash fingerprint is stored in the index. When all genomes have been inserted, each column can be compressed (gzip).
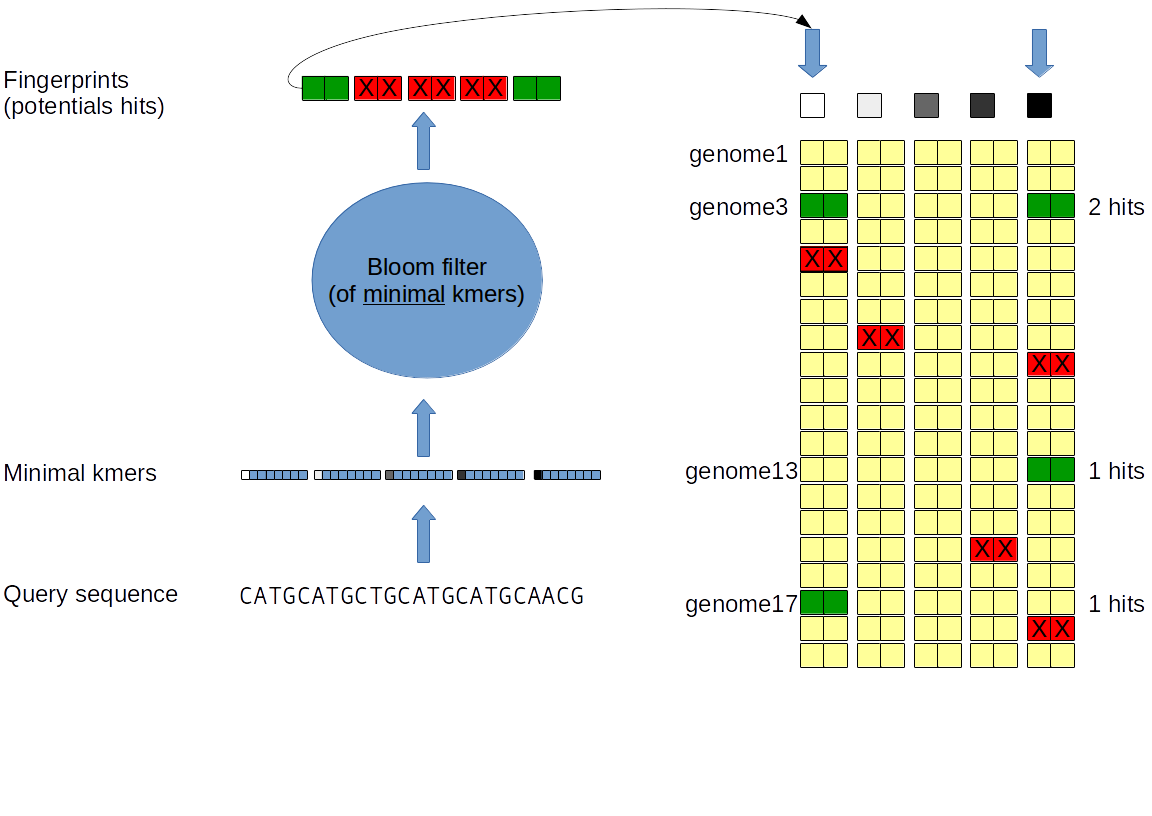
Index interrogation: The sketch of the sequence is computed. For each non empty partition: If the minimal kmer is in the Bloom filter: The fingerprints will be compared with the corresponding fingerprint column and genomes with the same fingerprint are accounted. The genomes sharing more than a given of fingerprint will be output. The union cardinality is estimated using the hyperloglog technique and the jaccard distance is estimated from the hit amount. The intersection cardinality is therefore deduced and output.
Three kinds of queries can be distinguished:
-
A large sequence (i.e. a genome) is queried. Most partition will be non-empty. If the query genome is similar to the indexed genomes, most column will be read.
-
A small sequence (i.e. a gene) is queried. Only a small fraction of the partition will be non empty. (A query composed of 1000 kmers will have a maximum a 1000 non empty partition). Therefore, only a low ammount of column will be read. For example a kilobase query will only need to read at most 1000 columns wich account for a small fraction of the index (100k to 1M columns). This is therefore an important optimization when is index is very large.
-
A unrelated (or erroneous) sequence is queried. Only a small fraction of the selected kmer will found in the Bloom filter. As before only relevant sub parts of the index will be read and compared. It also efficiently reduce the amount of False positive fingerprint matches that could occur "by chance".


Benchmark
Coming soon
Related works
Ultrafast search of all deposited bacterial and viral genomic data. Phelim Bradley, Henk C. den Bakker, Eduardo P. C. Rocha, Gil McVean and Zamin Iqbal
COBS: a Compact Bit-Sliced Signature Index Timo Bingmann, Phelim Bradley, Florian Gauger, and Zamin Iqbal
Fast search of thousands of short-read sequencing experiments B Solomon, C Kingsford
Dashing: Fast and Accurate Genomic Distances with HyperLogLog DN Baker, B Langmead
Mash: fast genome and metagenome distance estimation using MinHash Brian D. Ondov, Todd J. Treangen, Páll Melsted, Adam B. Mallonee, Nicholas H. Bergman, Sergey Koren and Adam M. Phillippy
HyperMinHash: MinHash in LogLog space Yun William Yu, Griffin M. Weber
Mash Screen: High-throughput sequence containment estimation for genome discovery Brian D Ondov, Gabriel J Starrett, Anna Sappington, Aleksandra Kostic, Sergey Koren, Christopher B Buck and Adam M Phillippy
A Memory-Efficient Sketch Method for Estimating High Similarities in Streaming Sets Pinghui Wang, Yiyan Qi, Yuanming Zhang, Qiaozhu Zhai, Chenxu Wang, John C.S. Lui, Xiaohong Guan
A Shifting Bloom Filter Framework for Set Queries T. Yang, A. Liu, M. Shahzad, Y. Zhong, Q. Fu, Z. Li