ACEP
Open source software and datasets for the ACEP algorithm
1.Making predictions for sequences
1.1 Description
ACEP is a deep learning model for high-throughput antibacterial peptide recognition. By loading a pre-trained model, the software can be deployed on a common computer to perform antimicrobial peptide recognition, important motif discovery, and visualization analysis. Both the peptide sequences and PSSM profiles need to be input the model.
1.2 Getting PSSM profiles
There are two ways to obtain PSSMs, a web servers or a standalone BLAST program. If your computer's memory is large enough (about 64GB), then you can run the standalone BLAST as described in section 1.2.2, otherwise we recommend to use an online server to obtain PSSMs.
1.2.1 Getting PSSM through online server
PSSM profiles of sequences can be obtained through POSSUM website (http://possum.erc.monash.edu/).
On POSSUM server page, the parameters of descriptors are all set to 'off' and the parameters for BLAST are set to defaults. After submitting sequences, wait for the server to finish calculation and download original PSSM profiles.
Note: if you want to use POSSUM online service, you have to fill the short sequence less than 50AA to more than 50AA in length by repeatedly copying the sequence, due to server restrictions on sequence length. (Or splicing signal peptide and propeptide to sequences to extend them.)
1.2.2 Getting PSSM through standalone BLAST program
-
Step 1. Go to the NCBI website to download the BLAST program (ncbi-blast-2.10.1+-win64.exe, https://ftp.ncbi.nlm.nih.gov/blast/executables/LATEST/).
Go to UniProt's website to download protein sequences (UniRef90 or UniRef50, fasta format, https://www.uniprot.org/downloads).
Unzip uniref90.fasta.gz to the bin folder of the ncbi-blast-2.10.1+-win64 installation directory (eg. E:\blast-2.9.0+\bin). -
Step 2. Use command line to enter the E:\blast-2.9.0\bin folder, run the following command to establish a local BLAST database.
makeblastdb -in uniref90.fasta -out uniref90.db -dbtype prot -
Step 3. Put a single sequence into the queryseq.fasta file, run the following command to generate a single PSSM 0001.pssm.
psiblast -db uniref90.db -query queryseq.fasta -out result1.out -out_ascii_pssm 0001.pssm -num_iterations 3 -evalue 1e-3 -
Step 4. If you need to generate PSSM profiles for a large number of sequences, you can store all the sequences in the queryseq.fasta file, and then execute ACEP/ACME_codes/localBLASTgetPSSMs/localBLASTgetPSSMs.py.
Three parameters need to be set in the localBLASTgetPSSMs.py program.
path0: the path of psiblast program (eg. path0 ='E:/blast-2.9.0/bin/psiblast').
path1: the path of BLAST database (eg. path1 ='E:/blast-2.9.0 /bin/uniref90.db').
path2: the path where the localBLASTgetPSSMs.py program is located (eg. path2 ='E:/ACEP/ACME_codes/localBLASTgetPSSMs/').
1.3 Requirements
Python3 Python packages: Tensorfollow(vr.1.6.0), Keras(vr.2.15), Matplotlib, scikit-learn, numpy, pandas and senborn(The senborn package is used for visualization).
We recommend using a GPU to speed up the calculations; if you use GPU acceleration, you also need to install cuda and cudnn.
1.4 Starting a prediction
-
Step 1. Download all the files in ACME_codes folder.
-
Step 2. Enter sequences into the 'AMP_prediction/inputs/sequences.csv' file and put the PSSM files into the 'AMP_prediction/inputs/PSSM_files/' directory.
Note: In order to increase the speed of high-throughput sequence prediction without generating additional errors, the PSSM files placed in the directory can only be named using numbers, such as 00001.pssm, 00002.pssm, 00003.pssm ..., and the order of these files that are sorted by file names must be the same as the order of sequences in the sequences.csv file. The refun() function in Data_pre_processing.py file will help to do this.
-
Step 3. Run ACEP_prediction.py.
-
Step 4. View predicted results in 'AMP_prediction/outputs/outputs.csv' file. And sequences with probability greater than or equal to 0.5 are identified as AMPs, and sequences with probability less than 0.5 are identified as non-AMPs.
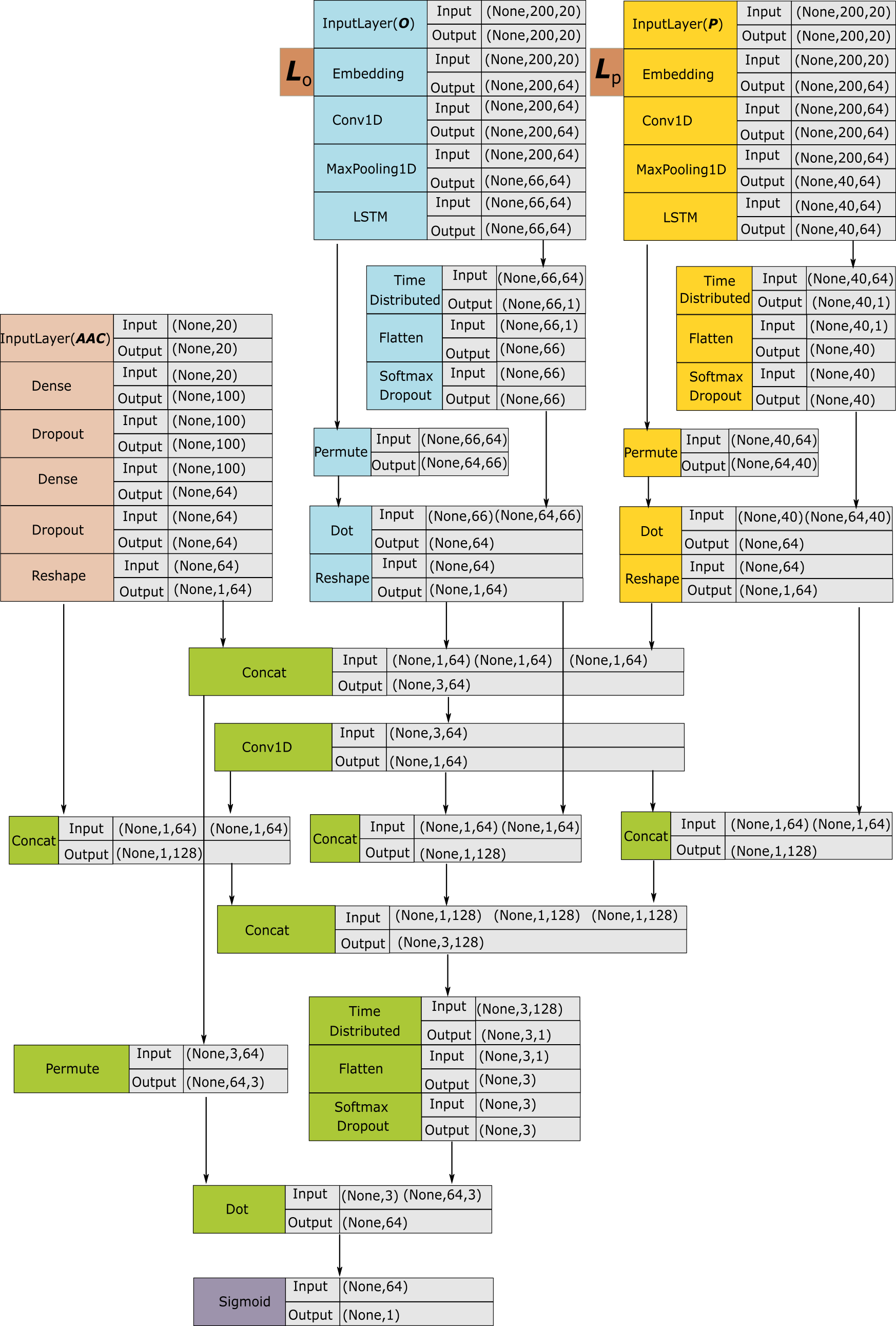
2.Architecture of the ACEP modle.
We use convolutional layer, pooling layer, LSTM layer, fully connected layer and attention mechanism to build the model. The yellow module, the blue module and the red module are used to generate features. The green module is used to fuse features; the purple module corresponds to sigmoid node that outputs the prediction results.
3.Repeating experiments in the paper.
Experiments in the paper can be repeated by running the code in the ACME_codes/ folder. When running the codes, all experimental results will be displayed and stored in the 'ACME_codes/experiment_results/ folder'.
3.1 Training model
You can train a new model by running ACEP_model_train.py files. And the code for the amino acid embedding tensor can be found in the EmbeddingRST.py file. The training data is placed in the AMPs_Experiment_Dataset / AMPs_Experiment_Dataset.zip file. This file needs to be decompressed before training the model. In addition, you can observe the training history by running the ACEP_training_history.py file.
3.2 Experimental comparison
You can view the performance of the model by running ACEP_model_performance.py. And it can also be compared with other state-of-the-art antimicrobial peptides recognition methods by running ACEP_comparison_test.py and ACEP_ROC.py. In addition, run ACEP_R1_xxx.py to understand the role of different functional modules. And evaluate model performance using cross validation on all datasets by running ACEP_model_CV.py
3.3 Experimental analysis and visualization
Observe amino acid clustering by running ACEP_cluster.py. Observe attention scores and motifs by running ACEP_attention_scores.py and ACEP_attention_motif.py. Observe the distribution of fusion features in space by running ACEP_fusion_feature.py.
3.4 Others
If your sequence is a '.fasta' file, you can call the function in Data_pre_processing.py to convert the fasta file to a '.csv' file so that it can be imported into the model. If you want to see false negative sequences, you can run ACEP_false_negtive.py file.
Contact Us
If you have any problems, please contact fhy11235813@gmail.com
Haoyi Fu
School of Information Science and Engineering, Yunnan University
Cite
If these codes are helpful to you, please cite our paper:
Fu, H., Cao, Z., Li, M. et al. ACEP: improving antimicrobial peptides recognition through automatic feature fusion and amino acid embedding. BMC Genomics 21, 597 (2020). https://doi.org/10.1186/s12864-020-06978-0
https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-06978-0