- Trio
yak count -b37 -t16 -o pat.yak <(cat pat_1.fq.gz pat_2.fq.gz) <(cat pat_1.fq.gz pat_2.fq.gz)
yak count -b37 -t16 -o mat.yak <(cat mat_1.fq.gz mat_2.fq.gz) <(cat mat_1.fq.gz mat_2.fq.gz)
hifiasm -o HG002.asm -t32 -1 pat.yak -2 mat.yak HG002-HiFi.fa.gz- HiC
hifiasm -o HG002.asm -t32 --h1 read1.fq.gz --h2 read2.fq.gz HG002-HiFi.fq.gz- Evaluation
## yak (https://github.com/lh3/yak)
yak trioeval pat.yak mat.yak HG002.asm.trio.hap1.p_ctg.fa > HG002.asm.hap1.trioeval
yak trioeval pat.yak mat.yak HG002.asm.trio.hap2.p_ctg.fa > HG002.asm.hap2.trioeval
## Merqury.FK (https://github.com/thegenemyers/MERQURY.FK)
- Remove Haplotype-specific HiC reads (https://www.nature.com/articles/s41586-021-03451-0)
meryl-lookup -memory 2 -exclude -mers pat.meryl -sequence $read1 -sequence2 $read2 -r2
mat.R2.fastq.gz | pigz -c > mat.R1.fastq.gz
meryl-lookup -memory 2 -exclude -mers mat.meryl -sequence $read1 -sequence2 $read2 -r2
pat.R2.fastq.gz | pigz -c > pat.R1.fastq.gz
Using https://github.com/marbl/merqury/blob/master/trio/exclude_reads.sh
bash /data/software/Merqury/merqury/trio/exclude_reads.sh mat.hapmer.meryl f1.hic.R1.fq.gz f1.hic.R2.fq.gz pat-
Scaffold two haplotype together and check (recommend)
-
run juicer + 3d-dna / AllHiC
-
Loading into JuiceBox for visualization
- False duplication (Examples are based on the hifiasm 0.15.4 for high het plant genome with trio data)
-
Fig1
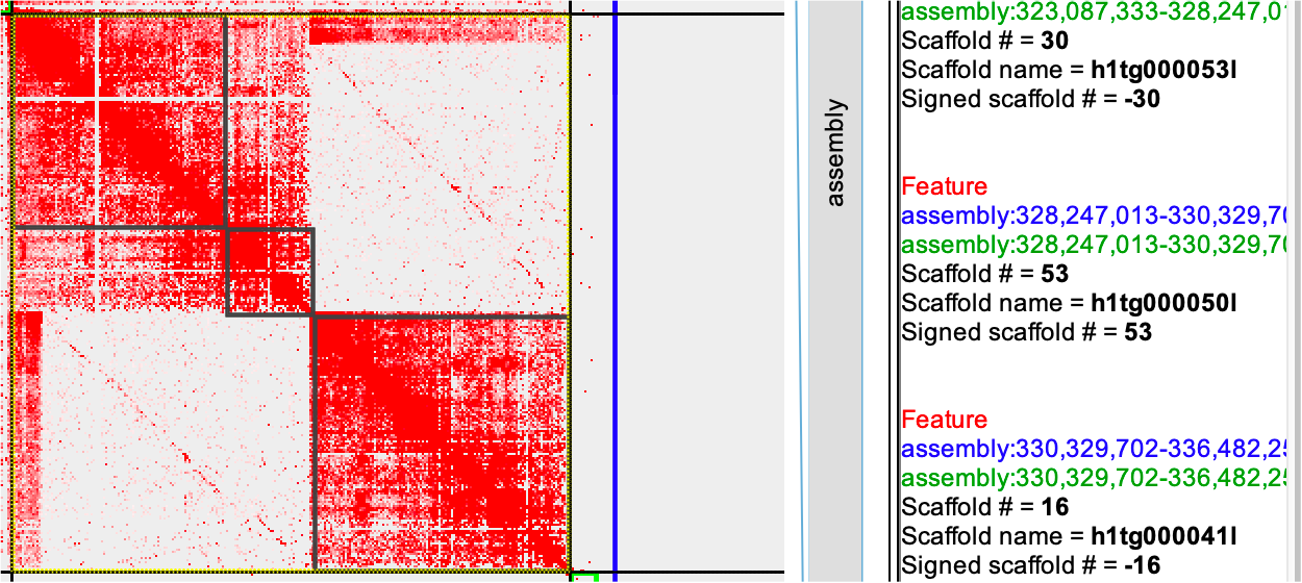
seqName #matKmer #patKmer #pat-pat #pat-mat #mat-pat #mat-mat seqLen h1tg000041l 160 10644 77 83 82 10561 6152553 h1tg000053l 50105 62 50059 45 45 17 5159680 h1tg000050l 17945 27 17924 20 20 7 2082689 Conclusion: Remove the h1tg000041l
-
Fig2
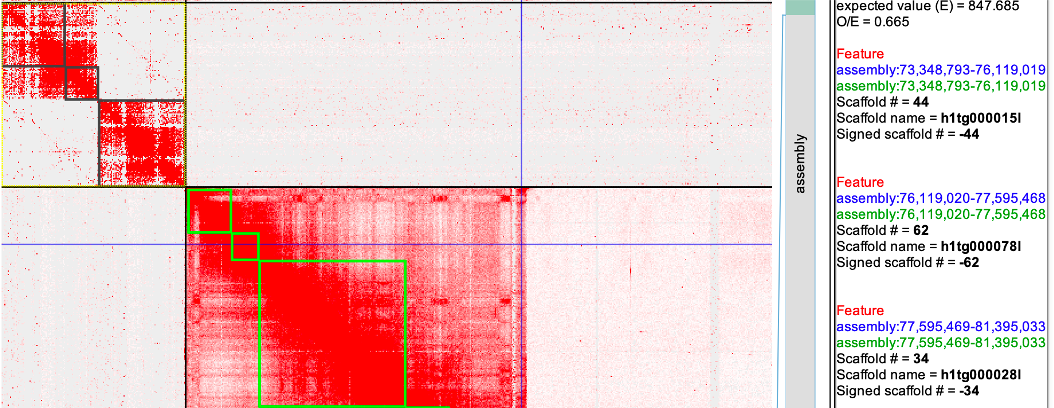
seqName #patKmer #patKmer #pat-pat #pat-mat #mat-pat #mat-mat seqLen h1tg000015l 87 164 51 35 36 128 2770227 h1tg000078l 42 66 24 18 18 47 1476449 h1tg000028l 7532 87 7478 53 53 34 3799565 Conclusion: Remove the h1tg000015l and h1tg000028l
-
Fig3
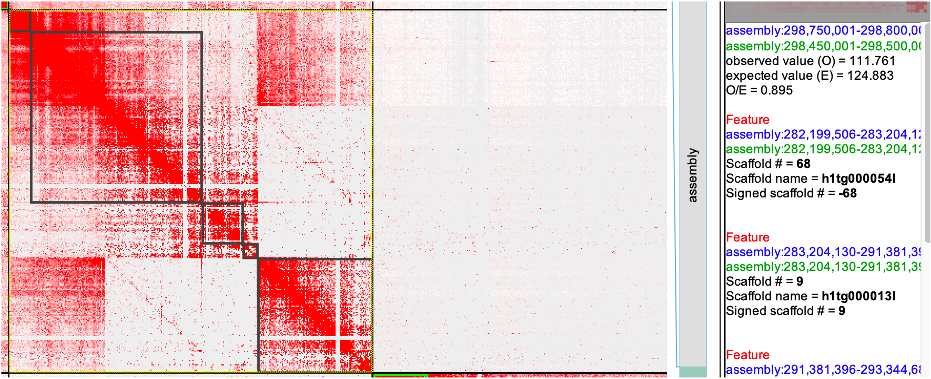
seqName #patKmer #patKmer #pat-pat #pat-mat #mat-pat #mat-mat seqLen h1tg000054l 6908 10 6899 8 8 2 1004624 h1tg000013l 25643 330 25547 95 96 234 8177266 h1tg000059l 57 245 25 32 32 212 1963293 h1tg000056l 12 29 4 7 7 22 685156 h1tg000052l 25905 219 25816 88 88 131 5467035 Conclusion: Remove the h1tg000059l and h1tg000056l, 52l vs (54l+13l) maybe real
-
Fig4
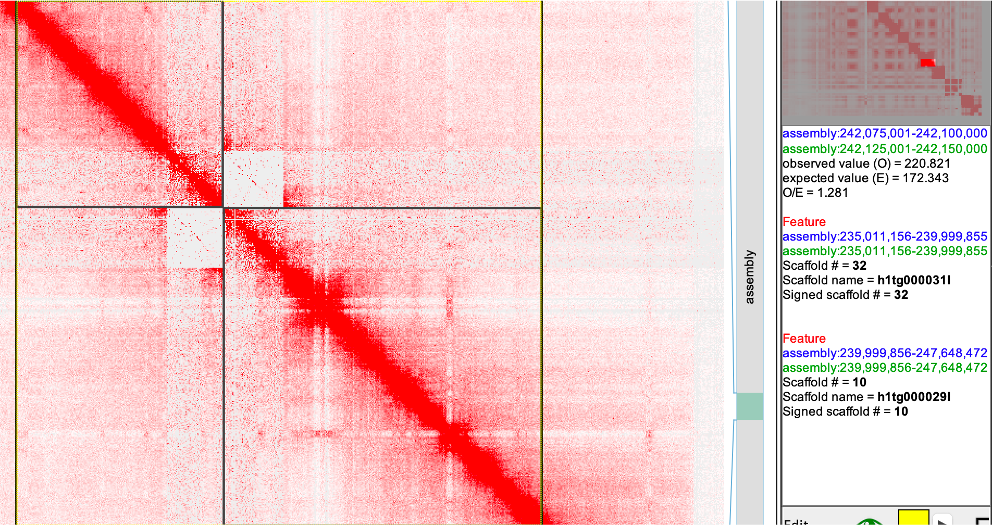
seqName #patKmer #patKmer #pat-pat #pat-mat #mat-pat #mat-mat seqLen h1tg000031l 17138 12244 17075 63 63 12180 4988700 h1tg000029l 44167 139 44067 99 99 40 7648617 Conclusion: utg overlap. Find the utg in the
gfaand remove rerun theutgtrioeval and decide. -
Fig5
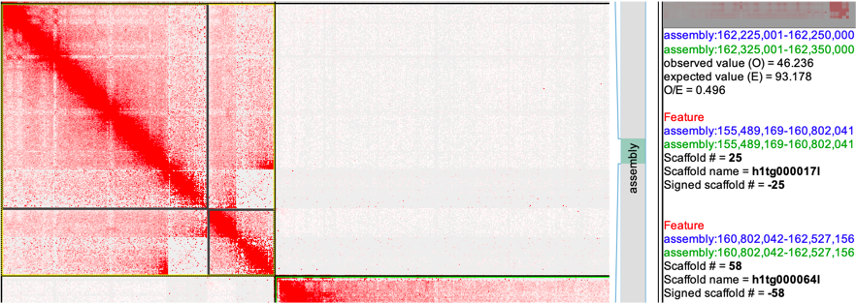
seqName #patKmer #patKmer #pat-pat #pat-mat #mat-pat #mat-mat seqLen h1tg000017l 39991 89 39929 61 61 28 5312873 h1tg000064l 1572 5187 1540 31 32 5155 1725115 Conclusion: utg overlap. Find the utg in the gfa and remove.
-
- False duplication (Examples are based on the hifiasm 0.15.4 for high het plant genome with trio data)
-
Misplaced haplotype If you find the
hap1ctg have more strong interaction with the hap2, andtrioevalsupport it, you need manually movethis hap1ctg to hap2 -
Haplotype-specific inversion
MQ>1filter for haplotype interaction, kepp the heatmap inside the haplotype is normal.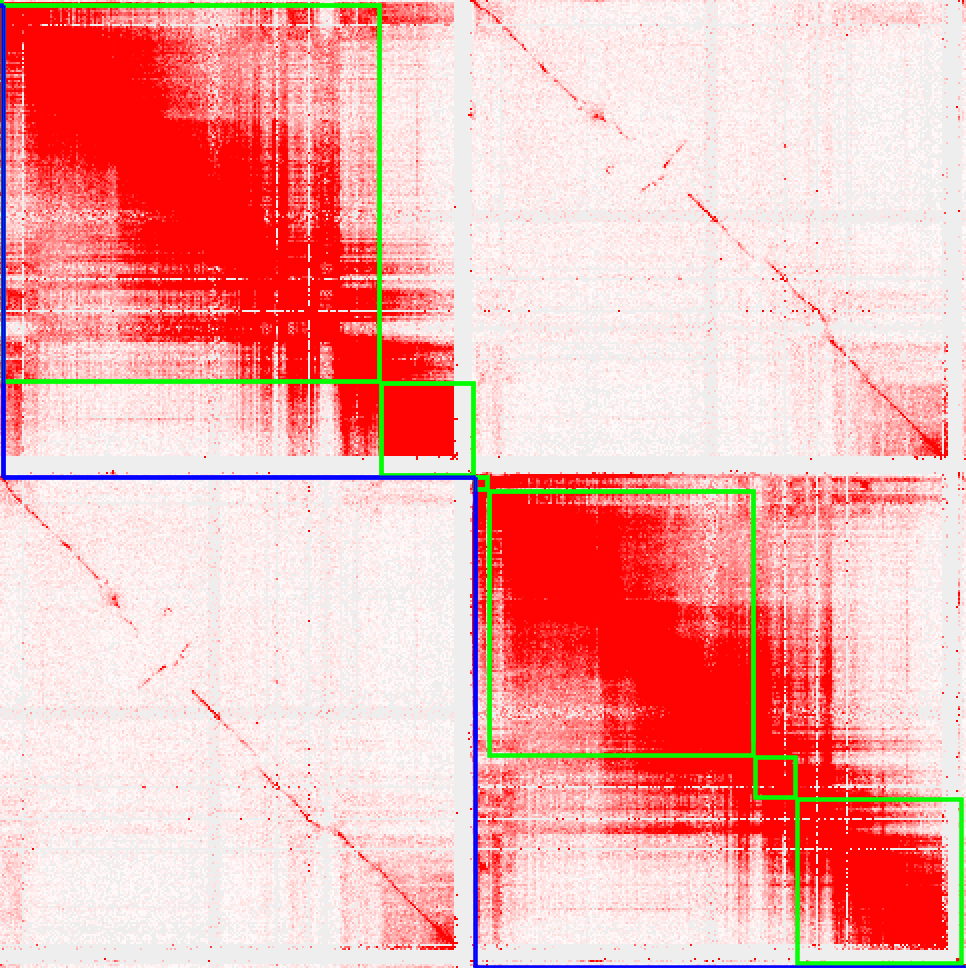
-






- Trioeval for hamming error and switch error
| Type | Hap | Switch | Hamming |
|---|---|---|---|
| Trio (0.14.2) | hap1 | 0.49% | 0.59% |
| Trio (0.14.2) | hap2 | 0.56% | 1.13% |
| Trio-check | hap1 | 0.44% | 0.39% |
| Trio-check | hap2 | 0.55% | 1.12% |
| Trio(0.16.1) | hap1 | 0.46% | 0.46% |
| Trio (0.16.1) | hap2 | 0.55% | 0.57% |
| HiC (0.16.1) | hap1 | 0.51% | 0.46% |
| HiC (0.16.1) | hap2 | 0.49% | 0.51% |
| dual (0.16.1) | hap1 | 0.51% | 8.51% |
| dual (0.16.1) | hap2 | 0.50% | 13.69% |
- QV estimation for two haplotype
- Check the haplotype synteny and confirm the utg (chhylp123/hifiasm#159)
- Check the coverage of two haplotype