The qPrimer package is a collection of tools for designing, checking, annotating, and visualizing qPCR primers.
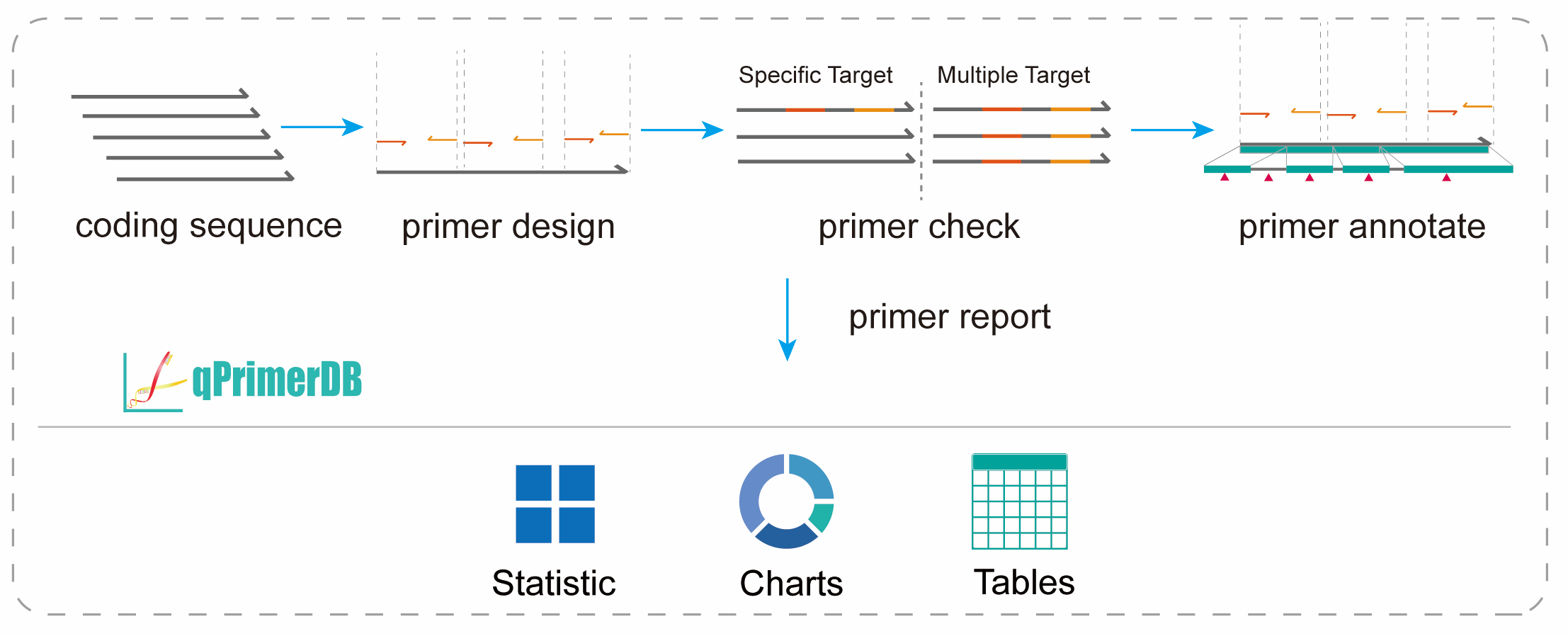
- In-Silico PCR (isPcr): A tool for checking primer specificity
- Python 3.9+ and the following packages:
- biopython: A set of tools for biological computation
- pandas: A fast, powerful, flexible, and easy-to-use open-source data analysis and data manipulation library
- numpy: The fundamental package for scientific computing with Python
- primer3-py: A Python wrapper for the Primer3 library
- jinja2: A fast, expressive, extensible templating engine
# install the package from source
git clone https://github.com/swu1019lab/qPrimer.git
cd qPrimer
# install the package using build and pip commands
pip install build --user
python -m build
pip install dist/qprimer-1.0.6.tar.gz --user# Display help information
qPrimer --helpusage: qPrimer [-h] [--version] {design,annotate,check,visualize} ...
Run qPrimer package.
positional arguments:
{design,annotate,check,visualize}
Specify the module to run
design Run design module
annotate Run annotate module
check Run check module
visualize Run visualize module
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exitusage: qPrimer design [-h] --seq_file SEQ_FILE [--ini_file INI_FILE] [--processes PROCESSES] [--csv] [--log_file LOG_FILE]
[--out_name OUT_NAME]
optional arguments:
-h, --help show this help message and exit
--seq_file SEQ_FILE Path to the sequence file with fasta format
--ini_file INI_FILE Path to the Primer3 configuration file with ini format
--processes PROCESSES
Number of processes to use
--csv Save the results to a CSV file
--log_file LOG_FILE Path to the log file
--out_name OUT_NAME Prefix name of the output filedefault parameters setting (can ignore upper case and lower case) in the ini file:
[GLOBAL]
primer_task = generic
primer_num_return = 10
primer_pick_left_primer = 1
primer_pick_right_primer = 1
primer_min_gc = 40
primer_max_gc = 60
primer_opt_gc_percent = 50
primer_opt_size = 22
primer_min_size = 18
primer_max_size = 28
primer_opt_tm = 60
primer_min_tm = 58
primer_max_tm = 64
primer_pair_max_diff_tm = 3
primer_product_size_range = 80-300
primer_explain_flag = 1
primer_thermodynamic_oligo_alignment = 1
PRIMER_SECONDARY_STRUCTURE_ALIGNMENT = 1
primer_annealing_temp = 50
PRIMER_MAX_POLY_X = 5
PRIMER_MAX_SELF_ANY = 4
PRIMER_MAX_SELF_END = 3
PRIMER_PAIR_MAX_COMPL_ANY = 4
PRIMER_PAIR_MAX_COMPL_END = 3
PRIMER_MAX_END_STABILITY = 9
[SEQUENCE]
[PROGRAM]Parameters specification:
- primer_task: generic
- primer_num_return: 10, number of primer pairs to return
- primer_pick_left_primer: 1, whether to pick the left primer, 0 or 1
- primer_pick_right_primer: 1, whether to pick the right primer, 0 or 1
- primer_min_gc: 40, minimum GC content, 0-100
- primer_max_gc: 60, maximum GC content, 0-100
- primer_opt_gc_percent: 50, optimal GC content, 0-100
- primer_opt_size: 22, optimal primer length
- primer_min_size: 18, minimum primer length
- primer_max_size: 28, maximum primer length
- primer_opt_tm: 60, optimal melting temperature
- primer_min_tm: 58, minimum melting temperature
- primer_max_tm: 64, maximum melting temperature
- primer_pair_max_diff_tm: 3, maximum difference in melting temperature between the left and right primers
- primer_product_size_range: 80-300, range of product size, e.g., 80-300 or 100-200, 300-400
- primer_explain_flag: 1, whether to include the explanation of the primer
- primer_thermodynamic_oligo_alignment: 1, whether to make thermodynamic secondary structure calculations
- primer_secondary_structure_alignment: 1, whether to print out the calculated secondary structures
- primer_annealing_temp: 50, annealing temperature
- primer_max_poly_x: 5, maximum number of consecutive G/C/T/A in the primer
- primer_max_self_any: 4, maximum self-complementarity
- primer_max_self_end: 3, maximum self-complementarity at the 3' end
- primer_pair_max_compl_any: 4, maximum complementarity between the left and right primers
- primer_pair_max_compl_end: 3, maximum complementarity between the 3' end of the left primer and the 3' end of the right primer
- primer_max_end_stability: 9, maximum stability of the 3' end of the primer
Parameters references:
- Jeon H, Bae J, Hwang SH, et al. MRPrimerW2: an enhanced tool for rapid design of valid high-quality primers with multiple search modes for qPCR experiments. Nucleic Acids Res. 2019;47(W1):W614-W622. doi:10.1093/nar/gkz323
- Lu K, Li T, He J, et al. qPrimerDB: a thermodynamics-based gene-specific qPCR primer database for 147 organisms. Nucleic Acids Res. 2018;46(D1):D1229-D1236. doi:10.1093/nar/gkx725
Custom parameters setting for qPCR and other applications:
All the parameters from Primer3 can be set in the ini file, and the default parameters are set for qPCR. The users can modify the parameters in the ini file to meet the specific requirements of the primer design not only for qPCR but also for other applications.
usage: qPrimer annotate [-h] --primers PRIMERS --gtf_file GTF_FILE --snp_file SNP_FILE [--processes PROCESSES] [--log_file LOG_FILE]
[--out_name OUT_NAME]
optional arguments:
-h, --help show this help message and exit
--primers PRIMERS Path to the primers results with json format
--gtf_file GTF_FILE Path to the GTF file
--snp_file SNP_FILE Path to the SNP file with bed format
--processes PROCESSES
Number of processes to use
--log_file LOG_FILE Path to the log file
--out_name OUT_NAME Prefix name of the output file
usage: qPrimer check [-h] --primers PRIMERS --database DATABASE [--processes PROCESSES] [--log_file LOG_FILE] [--out_name OUT_NAME]
optional arguments:
-h, --help show this help message and exit
--primers PRIMERS Path to the primers results with json format
--database DATABASE Path to the database file with fasta format
--processes PROCESSES
Number of processes to use
--log_file LOG_FILE Path to the log file
--out_name OUT_NAME Prefix name of the output fileusage: qPrimer visualize [-h] --primers PRIMERS --seq_file SEQ_FILE [--genes_num GENES_NUM] [--log_file LOG_FILE] [--out_name OUT_NAME]
optional arguments:
-h, --help show this help message and exit
--primers PRIMERS Path to the primers results with json format
--seq_file SEQ_FILE Path to the sequence file with fasta format
--genes_num GENES_NUM
Number of top genes to show
--log_file LOG_FILE Path to the log file
--out_name OUT_NAME Prefix name of the output fileThe default output format is JSON, and the output file is named qPrimer.json.
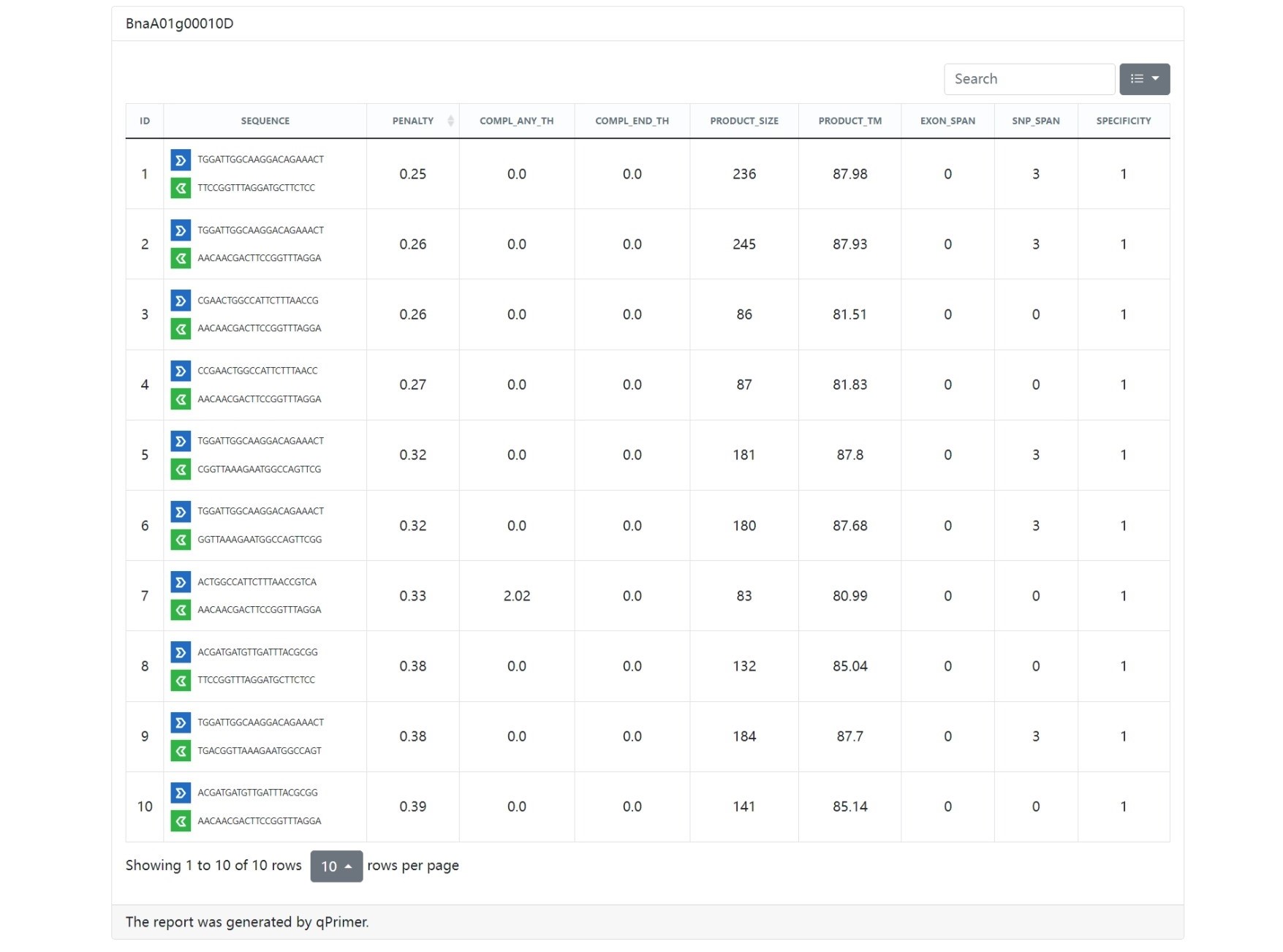
The output file contains the following three main sections:
-
Primer pair
- PENALTY: The penalty score of the primer pair
- PRODUCT_SIZE: The product size of the primer pair
- PRODUCT_TM: The product melting temperature of the primer pair
- COMPL_ANY_TH: The calculated value for the tendency of a primer pair to bind to each other
- COMPL_END_TH: The calculated value for the tendency of the 3'-ENDs of a primer pair to bind to each other.
-
Left primer
- PENALTY: The penalty score of the left primer
- SEQUENCE: The sequence of the left primer
- COORDS: The coordinates of the left primer
- TM: The melting temperature of the left primer
- BOUND: The fraction of primers bound at annealing temperature
- GC_PERCENT: The GC content of the left primer
- END_STABILITY: The stability of the 3' end of the left primer
- SELF_ANY_STUCT: A string representation of the calculated secondary structure
- SELF_END_STUCT: A string representation of the calculated secondary structure
- HAIRPIN_STUCT: A string representation of the calculated secondary structure
-
Right primer
- PENALTY: The penalty score of the right primer
- SEQUENCE: The sequence of the right primer
- COORDS: The coordinates of the right primer
- TM: The melting temperature of the right primer
- BOUND: The fraction of primers bound at annealing temperature
- GC_PERCENT: The GC content of the right primer
- END_STABILITY: The stability of the 3' end of the right primer
- SELF_ANY_STUCT: A string representation of the calculated secondary structure
- SELF_END_STUCT: A string representation of the calculated secondary structure
- HAIRPIN_STUCT: A string representation of the calculated secondary structure
In addition to the json output, the results can also be output in csv format with the --csv option.
The penalty score is used to evaluate the quality of the primer pair, and the primer pair with the lowest penalty score is considered the best primer pair.
The stability score is used to evaluate the stability of the 3' end of the primer, and the primer with the highest stability score is considered the most stable primer.
# Design primers with default json output
qPrimer design --seq_file test_cds.fa
# Design primers with json and csv output
qPrimer design --seq_file test_cds.fa --csv
# Design primers with multiple CPUs
qPrimer design --seq_file test_cds.fa --processes 4
# Design primers with specific parameters, like primer length, Tm, GC content, etc.
qPrimer design --seq_file test_cds.fa --ini_file test.ini
# Check primers specificity
qPrimer check \
--primers qPrimer.json \
--database test.fa
# Annotate primers with gene information and SNP information
qPrimer annotate \
--primers qPrimer.json \
--gtf_file genes.gtf \
--snp_file test_snps.bed
# Visualize primers results with a html page
qPrimer visualize \
--primers qPrimer.json \
--seq_file test_cds.fa \
--genes_num 10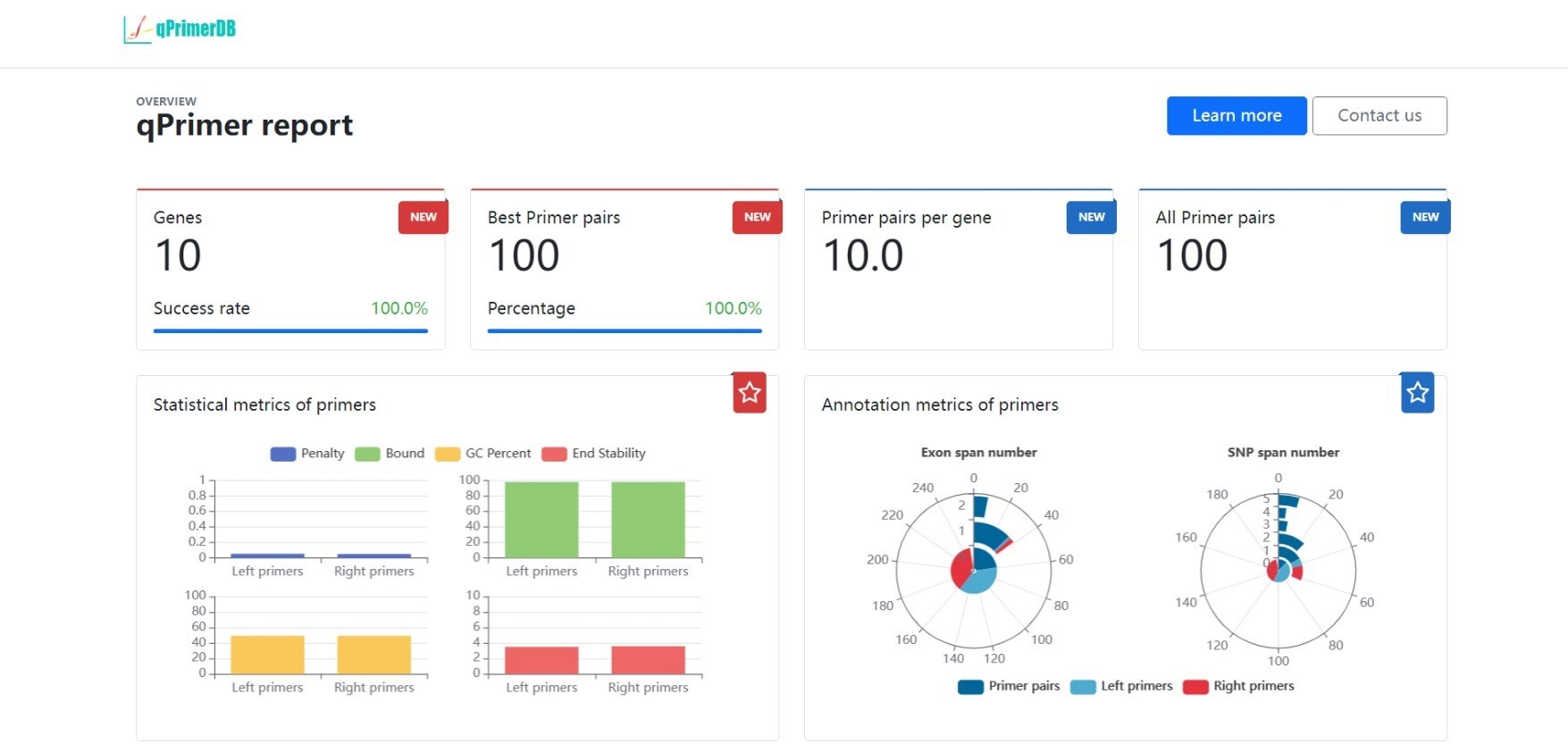
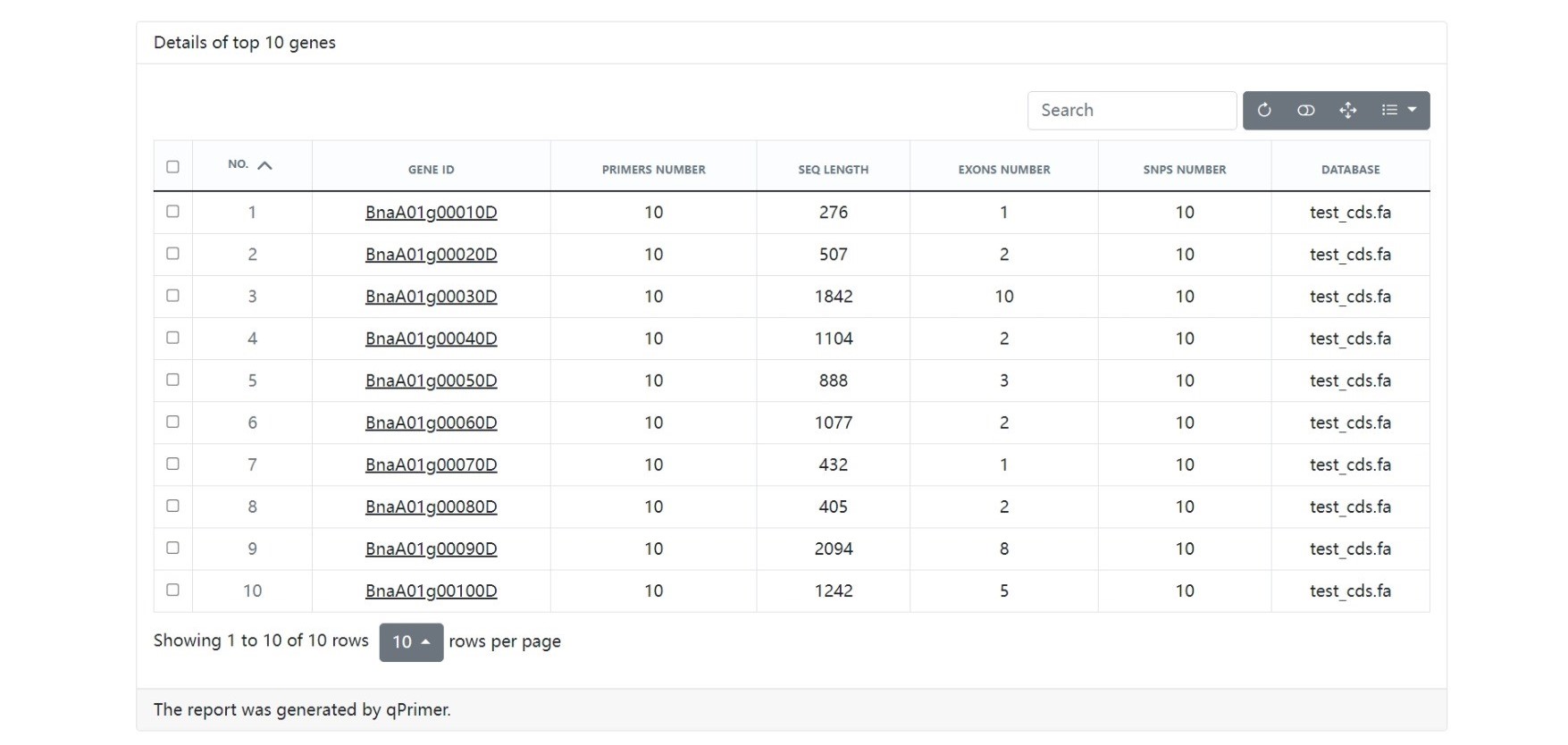
If you use qPrimerDB, please cite the following paper:
Lu K, Li T, He J, et al. qPrimerDB: a thermodynamics-based gene-specific qPCR primer database for 147 organisms. Nucleic Acids Res. 2018;46(D1):D1229-D1236. doi:10.1093/nar/gkx725
Li X, Meng B, Zhang Z, et al. qPrimerDB 2.0: an updated comprehensive gene-specific qPCR primer database for 1172 organisms. Nucleic Acids Res. Published online August 9, 2024. doi:10.1093/nar/gkae684
if you use qPrimer, please cite the following paper:
Li X, Meng B, Zhang Z, et al. qPrimerDB 2.0: an updated comprehensive gene-specific qPCR primer database for 1172 organisms. Nucleic Acids Res. Published online August 9, 2024. doi:10.1093/nar/gkae684
-
How to calculate the penalty values of the primer pair?
The penalty values define what is the best primer pair, and the lower the penalty score, the better the primer pair. the detailed calculation method can be found in the Primer3 manual. Simply, the penalty score is calculated by the sum of penalty weight of different parameters, such as the melting temperature, GC content, primer length, secondary structure, etc. Users can set the penalty weight of different parameters in the ini file.
-
How to calculate the stability values of the 3' end of the primer?
The value is the maximum delta G (kcal/mol) for duplex disruption for the five 3' bases as calculated using the nearest-neighbor parameter values specified by the option of melting temperature calculation. Bigger numbers mean more stable 3' ends. The detailed calculation method can be found in the Primer3 manual. Users can set the maximum stability value in the ini file.
-
How to calculate the secondary structure of the primer?
The secondary structure of the primer is calculated by the nearest-neighbor method. The detailed calculation method can be found in the Primer3 manual. Users can get the secondary structure of the primer by setting the parameter
primer_thermodynamic_oligo_alignmentto1in the ini file.