本项目是对2016年全国研究生数学建模竞赛B题的解答,原问题如下:
问题一、请用适当的方法,把genotype.dat中每个位点的碱基(A,T,C,G) 编码方式转化成数值编码方式,便于进行数据分析。
问题二、根据附录中1000个样本在某条有可能致病的染色体片段上的9445个位点的编码信息(见genotype.dat)和样本患有遗传疾病A的信息(见phenotype.txt文件)。设计或采用一个方法,找出某种疾病最有可能的一个或几个致病位点,并给出相关的理论依据。
问题三、同上题中的样本患有遗传疾病A的信息(phenotype.txt文件)。现有300个基因,每个基因所包含的位点名称见文件夹gene_info中的300个dat文件,每个dat文件列出了对应基因所包含的位点(位点信息见文件genotype.dat)。由于可以把基因理解为若干个位点组成的集合,遗传疾病与基因的关联性可以由基因中包含的位点的全集或其子集合表现出来请找出与疾病最有可能相关的一个或几个基因,并说明理由。
问题四、在问题二中,已知9445个位点,其编码信息见genotype.dat文件。在实际的研究中,科研人员往往把相关的性状或疾病看成一个整体,然后来探寻与它们相关的位点或基因。试根据multi_phenos.txt文件给出的1000个样本的10个相关联性状的信息及其9445个位点的编码信息(见genotype.dat),找出与multi_phenos.txt中10个性状有关联的位点。
本项目针对位点的特征选择问题,运用假设检验、模型选择、回归随机森林、逻辑回归、岭回归、多标签随机森林等方法对问题进行求解。
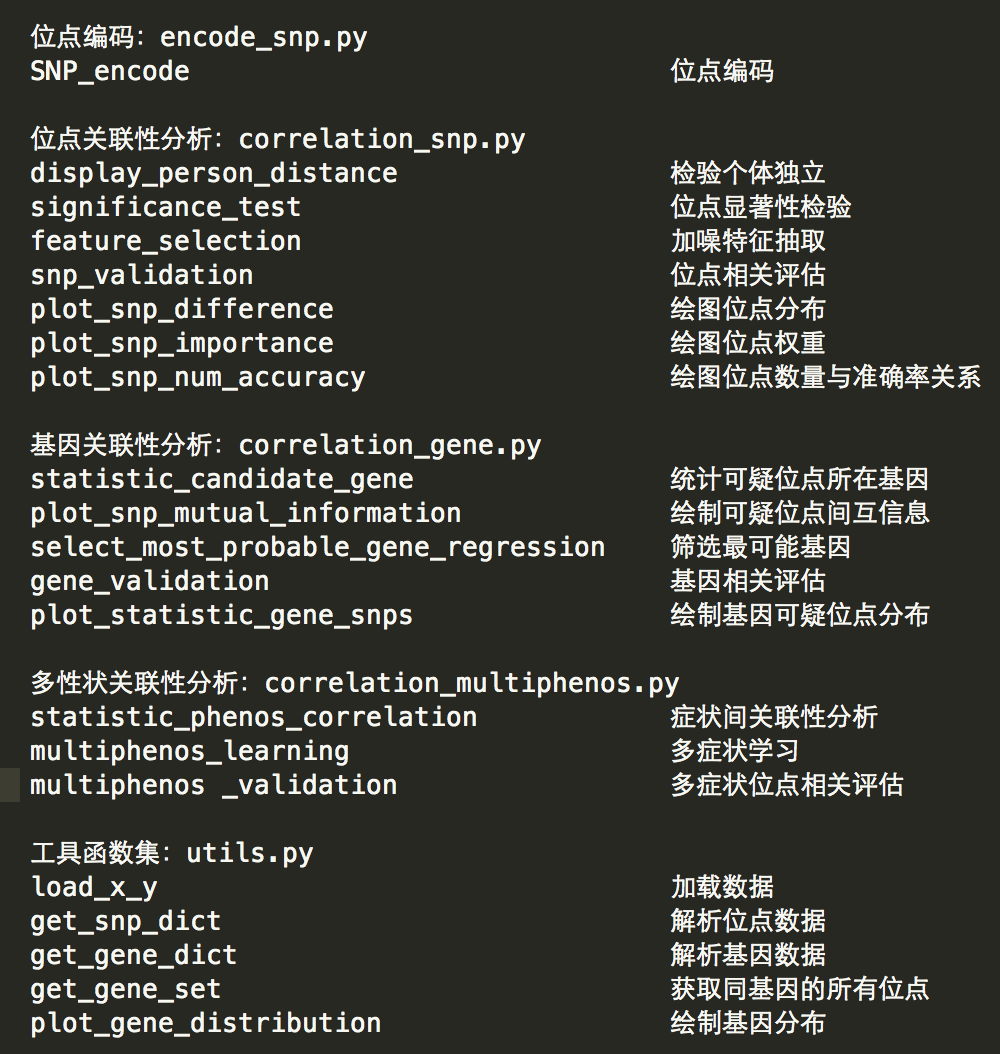
对于问题1,本项目根据分析需求,采用了两种编码方式:(1)基于位点碱基分布的{0,1,2}编码方式。(2)独热编码(One-hot)编码方式,分别用于解决假设检验和分类回归问题。
遗传位点碱基分布如图所示,可见,在rs7543405,rs12145450,rs214380位点中,由两种不同的碱基所组成的样本,患病概率最高。在这种场景下,采用独热编码的编码方式优于连续数值编码。
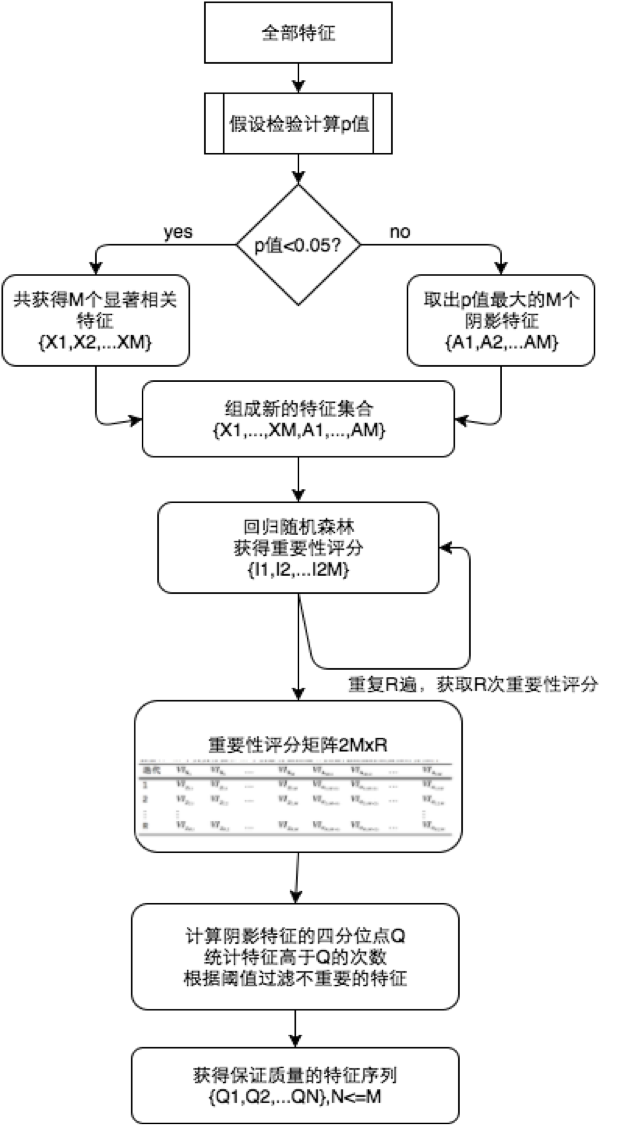
对于问题2,本项目提出了一种高维数据保证质量的特征选择模型,模型融合了特征选择问题算法的过滤式选择模型和包裹式选择模型,首先基于信息增益对所有特征进行特征的相关性评价,选出相关性最显著的特征作为候选特征,最无关的特征作为“阴影”特征,然后针对筛选过的特征采用回归随机森林进行变量重要性评价,通过迭代的方式过滤重要性低于阴影特征四分位点的候选特征,选出真正重要的特征。经过结果检验,可以发现本模型选择出了最重要的40个位点,10折交叉验证中对于遗传疾病A预测的准确率达到72%。
基于质量保证的特征选择算法流程图如下。
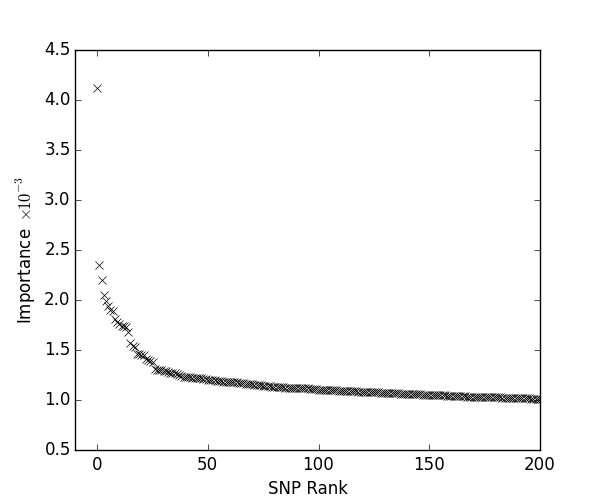
如图所示的是遗传位点重要性从大到小排名的曲线,可以看出,小部分遗传位点具有高的变量重要性,大部分遗传位点具有很低的变量重要性,可以解释为长尾效应。从图中可以看出,大约在排名40的位点,变量重要性骤减。
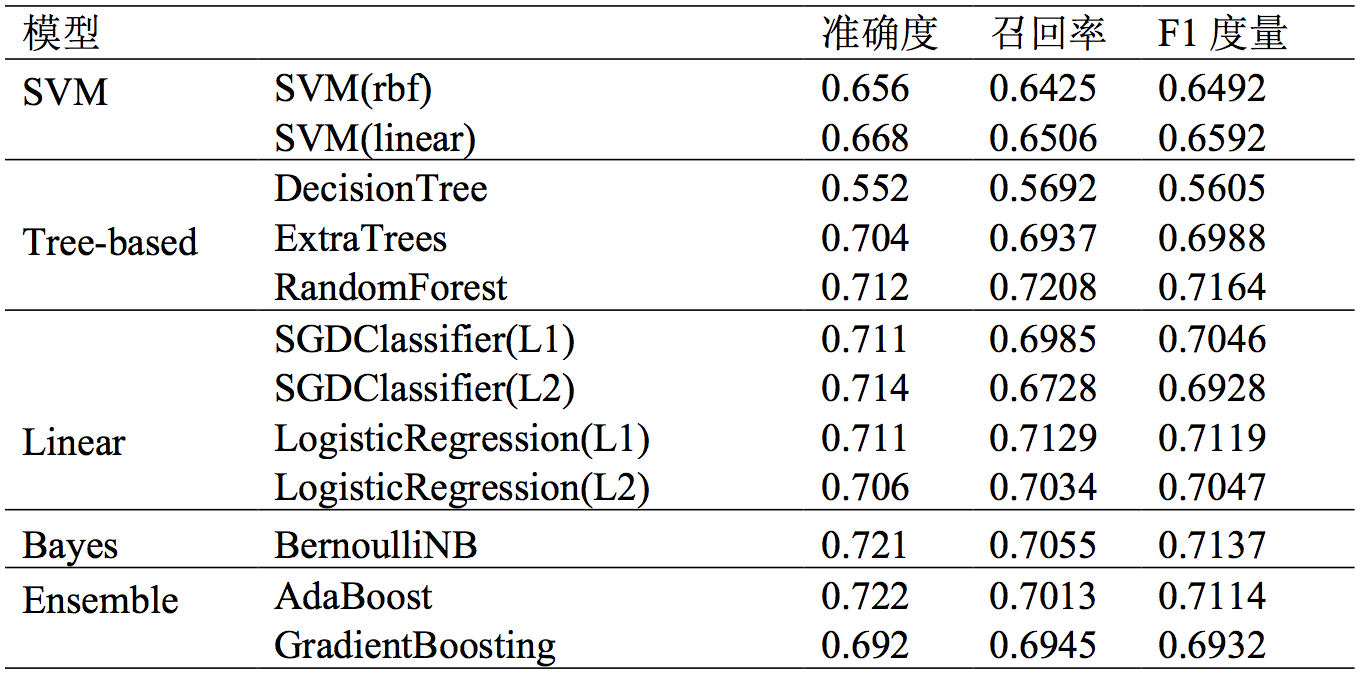
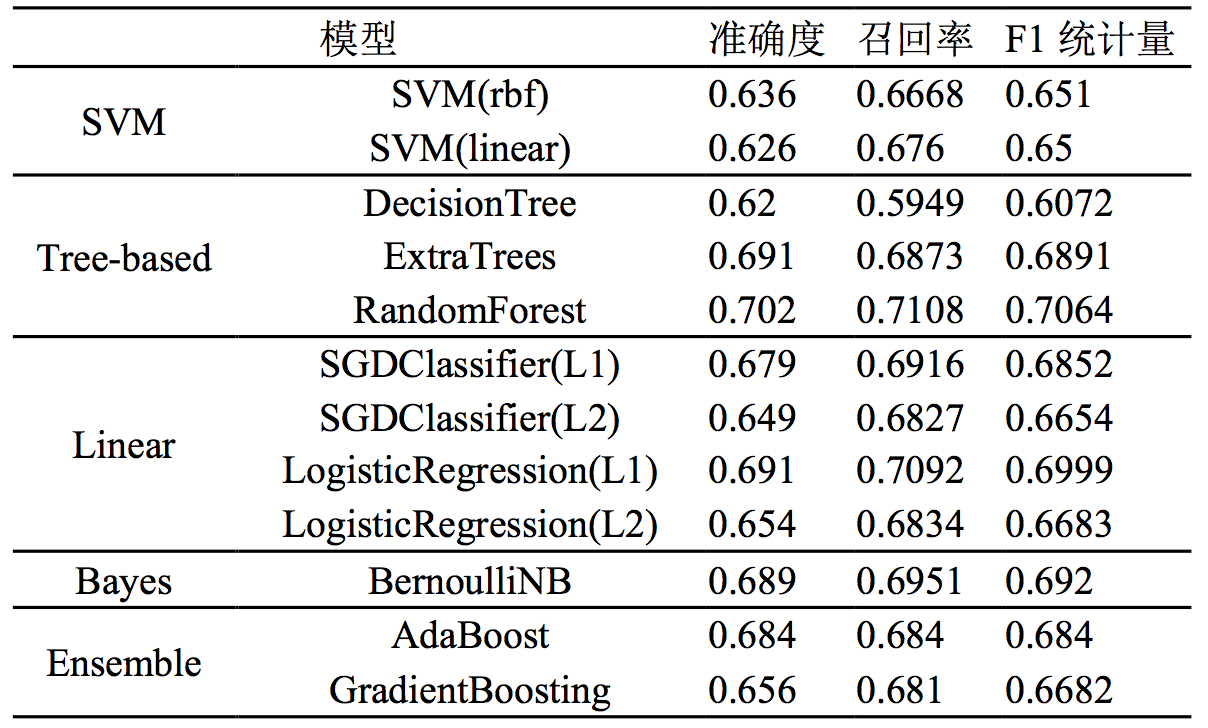
对于模型产生的结果,将产生的40个特征作为变量,对样本的健康状况进行预测。测试采用了支持向量机模型、基于树的模型、线性模型、贝叶斯模型和组合模型。因为原始数据集的数据量偏小,采用10折交叉验证的方法,各种模型的测试集准确率、召回率、F1度量如表所示。由表可见,随机森林模型对于预测疾病A的表现最优秀,F1度量达到了71.64%,AdaBoost的准确率最高,达到了72.2%。
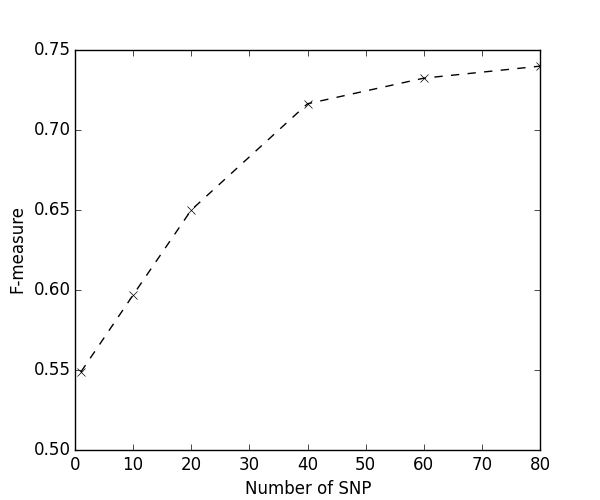
随着质量系数阈值的降低,可以获得更多的位点,从而达到更高的预测准确度,如图所示是F1度量与参与预测的位点数量的关系,可见,从1个到40位个点的过程中,F1度量快速上升,在40个输入特征时,达到72%。从40个到80个位点的过程中,F1度量缓慢上升最终在80个位点时,达到了74%。可以确认,保证质量的特征选择模型的确选出了真正重要的致病位点。
对于问题3,本项目首先采用互信息模型衡量了位点之间相关性,验证了相同基因的不同位点之间存在连锁不平衡性质,因此不能假设位点之间相互独立,之后采用了逻辑回归和岭回归分类模型分别对各个基因中的可疑位点和疾病之间进行回归分析,并基于决定系数挑选出了20个与疾病最相关的基因,这些基因通常包含较多的可疑位点或其中某个位点的重要性评分较高。经过结果检验,验证了获取的最可能的这20个基因对于遗传疾病A预测的准确率达到70%。
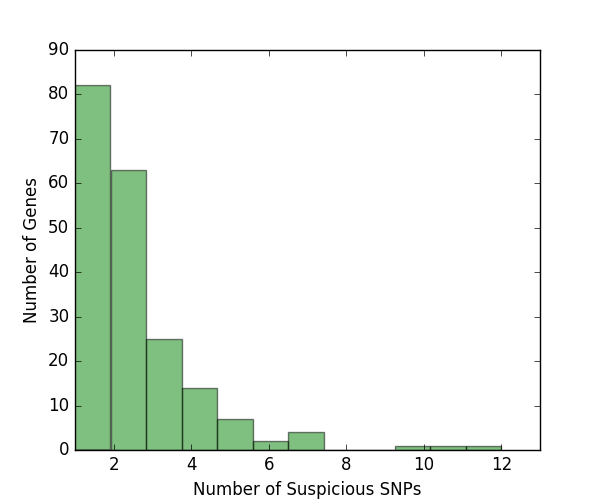
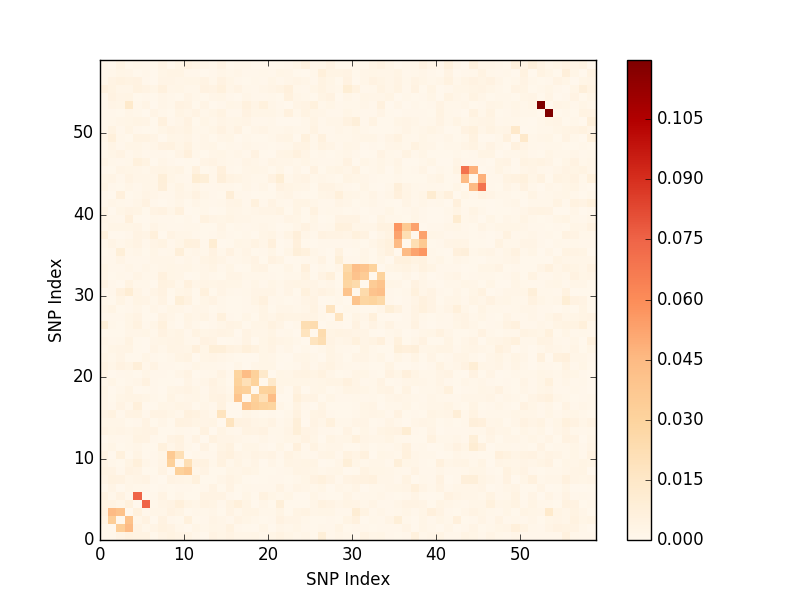
首先统计出了基因中包含可疑位点的数量的分布统计,分布如图所示,横坐标是基因中包含位点的数量,纵坐标是对应的基因数量,可见,大多数基因只包含少量位点,但仍然有相当比重的基因包含两个或以上的位点。
采用互信息模型,衡量了相同基因之间以及不同基因之间的位点的相关性,结果如图所示。其中横、纵坐标均是疑似位点按照原顺序(染色体上的顺序)进行排序,其中颜色较深部分的互信息较大,对应的位点之间可能存在某种关系,由结果的分布可知,对角线附近的位点存在互信息较大部分,而非对角线部分的互信息均较小,而角线附近的位点距离较近,经过验证除去极少数基因,互信息较大的基因均存在与相同的染色体上,验证了基因中位点存在连锁不平衡性质,因此不能假设基因中位点相互独立。
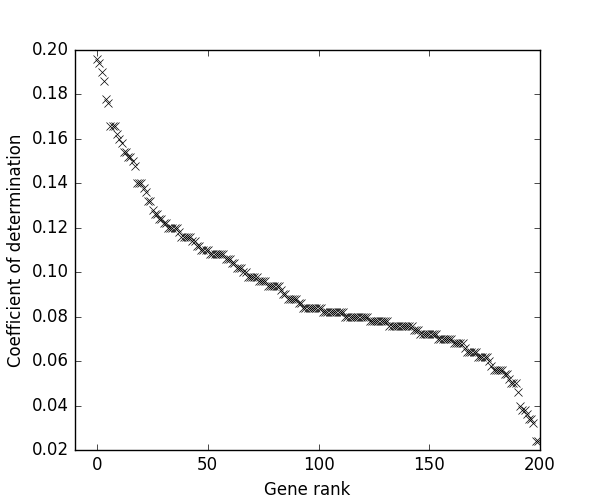
如图所示是对基因按照决定系数由大到小进行排序后的决定系数的分布,由此可见曲线存在两个拐点,其中左边第一个拐点靠左的基因决定系数较大,而往右决定系数变化趋缓,可以认为左边的20-40个是更有可能的基因集合。
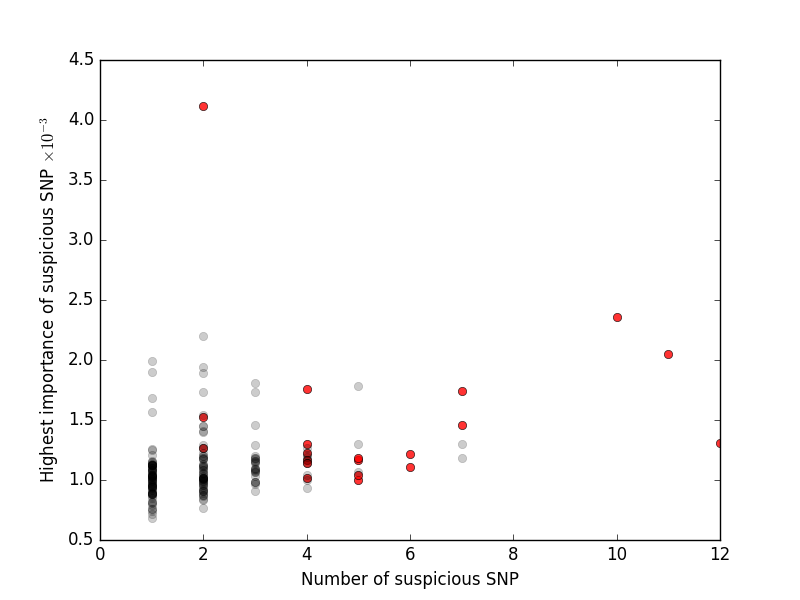
如图所示是对所选中的20个决定系数最大的基因的特性的分布,包含两个维度,即基因中包含可疑位点的数量和包含的可疑位点的重要性评分的最高分,说明包含可疑位点数量较多,或其中包含的可疑位点的重要性评分的最高分较高的基因更有可能和疾病相关,同时只包含一个可疑位点的基因均为被选中,一定概率避免了由于某种巧合因素所导致的假阳性,而同时也存在包含多个可疑位点而未被选中的情况,这可能是因为这些位点具有较高的互信息,回归系数不够显著。
最后通过与问题二相同的验证方法,验证了获取的最可能的20个基因为最有可能和疾病A相关的基因。采用10折交叉验证的方法,多种模型的测试集准确率、召回率、F1统计量如表所示。采用这些基因获取了趋近于问题二评估的准确率。
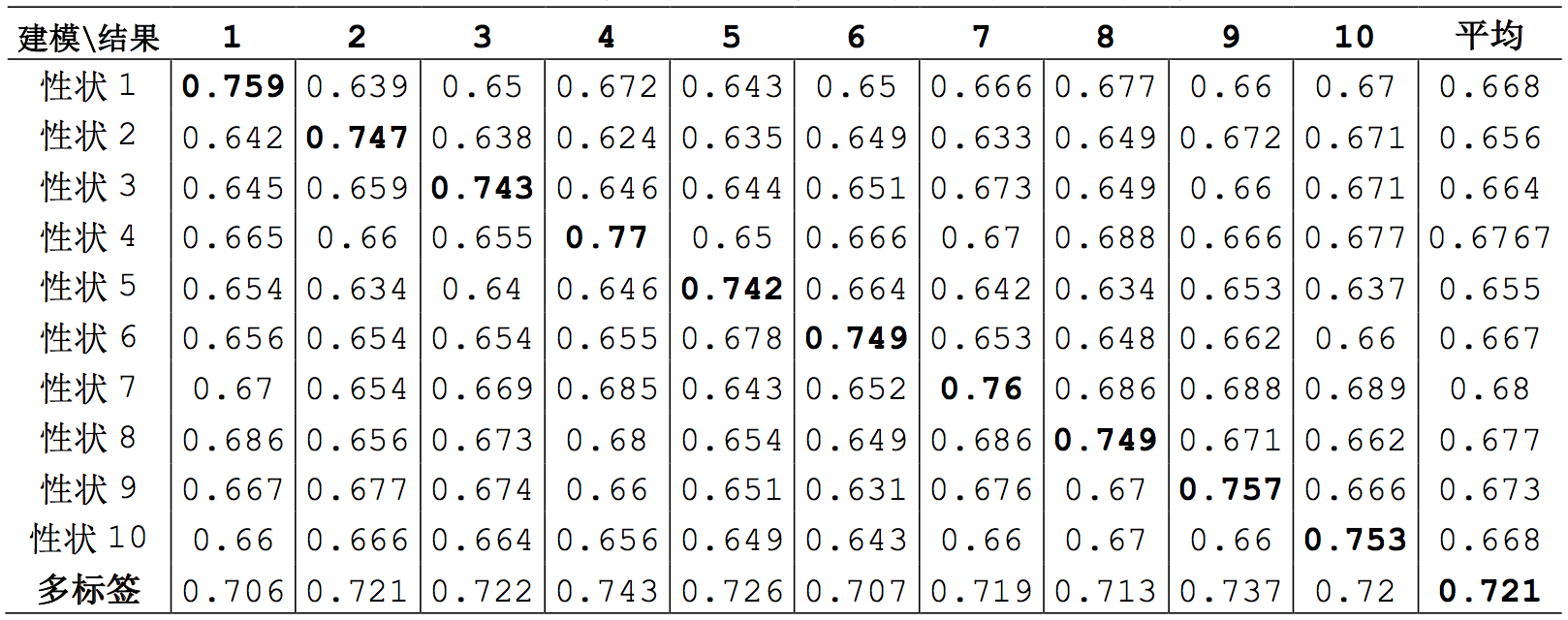
对于问题4,本项目根据多个位点对应多个性状这一特点本项目把它归为多标签分类问题(Multi-label Classification)建模。首先对性状之间的相关性作了分析,结合数据本身特点以及性状之间的强相关性,本项目选择算法适应法(Algorithm Adaptation),以Random Forest of ML-C4.5 (RFML-C4.5)建立模型。通过假设检验与多性状投票相结合的方式进行特征筛选,通过MLRF模型选择40个最重要特征,通过10折交叉验证得到72.1%的准确率,并通过对比试验证明了多标签模型较单标签模型的优势。
对于选出来的40个位点,通过RF模型进行10折交叉验证,以准确率(accuracy)作为评估指标。如表所示,最后一行是使用多标签模型的预测结果,可见多标签模型对10个性状的预测准确率都比较高,均为70%以上,10个性状准确率的平均值达72%。为了验证模型的性能,与单标签模型进行了对比。对比试验中,针对10个性状中的每一种性状,采用问题二中的解决方案,筛选出40个特征,之后使用这40个特征,采用随机森林模型,对每一个性状10折建模预测,得到相应的准确率。例如,使用性状1进行单模型性状选择,得到了40个性状,然后使用这40个性状对10个性状分别做10折预测。观察表第一行可见,通过性状1选出的40个性状,对自身预测准确率高达75.9%,但对其他9个性状预测准确率较低,10个性状准确率平均值较低(66.8%)。其他性状的单标签预测也类似。可见,通过多标签分类模型选出的位点集合,与所有的性状整体相关度最高;而通过单标签分类模型选出位点集合,仅仅与其中的某个性状相关性较高,而与其他性状的相关性较低。