



- Introduction
- Installation
- Testing
- Inputs
- Outputs
- EDTA usage
- Benchmarking usage
- Citations
- Other resources
- Issues
- Acknowledgements
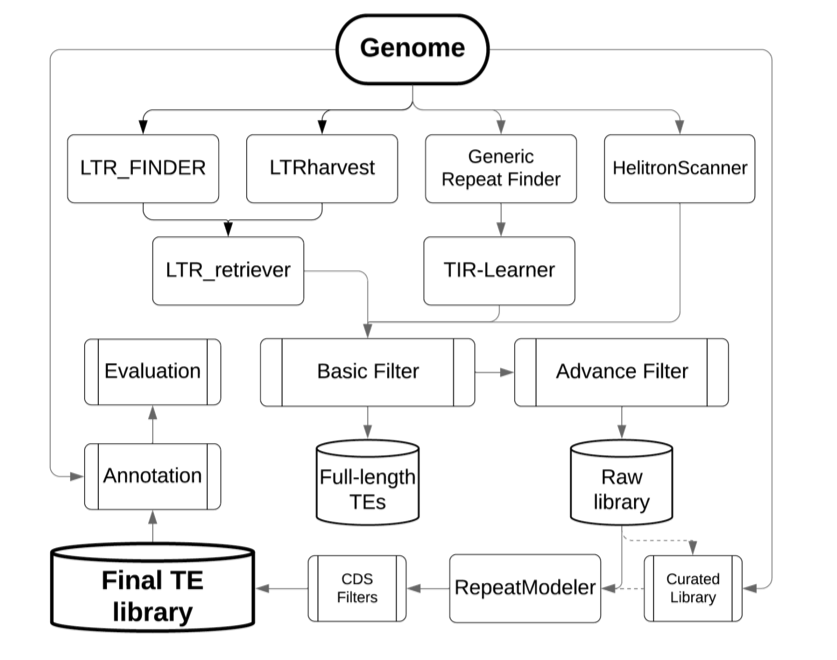
This package is developed for automated whole-genome de-novo TE annotation and benchmarking the annotation performance of TE libraries.
The EDTA package was designed to filter out false discoveries in raw TE candidates and generate a high-quality non-redundant TE library for whole-genome TE annotations. Selection of initial search programs were based on benckmarkings on the annotation performance using a manually curated TE library in the rice genome.
For benchmarking of a testing TE library, I have provided the curated TE annotation (v6.9.5) for the rice genome (TIGR7/MSU7 version). You may use the lib-test.pl script to compare the annotation performance of your method/library to the methods we have tested (usage shown below).
There are four ways to install EDTA. Please choose one.
conda install -c bioconda -c conda-forge edta
Quick installation using Singularity (good for HPC users)
Installation:
singularity pull EDTA.sif docker://quay.io/biocontainers/edta:<tag>
Visit BioContainers' Quay.io repository for a list of available tags.
Usage:
singularity exec {path}/EDTA.sif EDTA.pl --genome genome.fa [other parameters]
Where {path} is the path you build the EDTA singularity image
Quick installation using Docker (good for root/Mac users)
Installation:
docker pull quay.io/biocontainers/edta:<tag>
Usage:
docker run -v $PWD:/in -w /in biocontainers/edta:<tag> --genome genome.fa [other parameters]
Visit BioContainers' Quay.io repository for a list of available tags.
conda create -n EDTA
conda activate EDTA
conda config --env --add channels anaconda --add channels conda-forge --add channels bioconda
conda install -n EDTA -y cd-hit repeatmodeler muscle mdust blast openjdk perl perl-text-soundex multiprocess regex tensorflow=1.14.0 keras=2.2.4 scikit-learn=0.19.0 biopython pandas glob2 python=3.6 tesorter genericrepeatfinder genometools-genometools ltr_retriever ltr_finder numpy=1.16.4
git clone https://github.com/oushujun/EDTA
./EDTA/EDTA.pl
You can test the EDTA pipeline with a 1-Mb toy genome (it takes about 5 mins):
cd ./EDTA/test
perl ../EDTA.pl --genome genome.fa --cds genome.cds.fa --curatedlib ../database/rice6.9.5.liban --exclude genome.exclude.bed --overwrite 1 --sensitive 1 --anno 1 --evaluate 1 --threads 10
Required: The genome file [FASTA]. Please make sure sequence names are short (<=15 characters) and simple (i.e, letters, numbers, and underscore).
Optional:
- Coding sequence of the species or closely related species [FASTA]. This file helps to purge gene sequences in the TE library.
- Known gene position of this version of the genome assembly [BED]. Coordinates specified in this file will be whitelisted from TE annotation to avoid over-masking.
- Curated TE library of the species [FASTA]. This file is trusted 100%. Please make sure it's curated. If you only have a couple of curated sequences, that's fine. It doesn't need to be complete.
Expected: A non-redundant TE library: $genome.mod.EDTA.TElib.fa. The curated library is included in this file if provided. TEs are classified into the superfamily level and using the three-letter naming system reported in Wicker et al. (2007). Each sequence can be considered as a TE family.
Optional:
- Novel TE families: $genome.mod.EDTA.TElib.novel.fa. This file contains TE sequences that are not included in the curated library (
--curatedlibrequired). - Whole-genome TE annotation: $genome.mod.EDTA.TEanno.gff. This file contains both structurally intact and fragmented TE annotations (
--anno 1required). - Summary of whole-genome TE annotation: $genome.mod.EDTA.TEanno.sum (
--anno 1required). - Low-threshold TE masking: $genome.mod.MAKER.masked. This is a genome file with only long TEs (>=1 kb) being masked. You may use this for de novo gene annotations. Annotated gene models should contain TEs and need further filtering (
--anno 1required). - Annotation inconsistency for simple TEs; $genome.mod.EDTA.TE.fa.stat.redun.sum (
--evaluate 1required). - Annotation inconsistency for nested TEs: $genome.mod.EDTA.TE.fa.stat.nested.sum (
--evaluate 1required). - Oveall annotation inconsistency: $genome.mod.EDTA.TE.fa.stat.all.sum (
--evaluate 1required).
You got a genome and you want to get a high-quality TE annotation:
perl EDTA.pl [options]
--genome [File] The genome FASTA
--species [Rice|Maize|others] Specify the species for identification of TIR candidates. Default: others
--step [all|filter|final|anno] Specify which steps you want to run EDTA.
all: run the entire pipeline (default)
filter: start from raw TEs to the end.
final: start from filtered TEs to finalizing the run.
anno: perform whole-genome annotation/analysis after TE library construction.
--overwrite [0|1] If previous results are found, decide to overwrite (1, rerun) or not (0, default).
--cds [File] Provide a FASTA file containing the coding sequence (no introns, UTRs, nor TEs) of this genome or its close relative.
--curatedlib [file] Provided a curated library to keep consistant naming and classification for known TEs.
All TEs in this file will be trusted 100%, so please ONLY provide MANUALLY CURATED ones here.
This option is not mandatory. It's totally OK if no file is provided (default).
--sensitive [0|1] Use RepeatModeler to identify remaining TEs (1) or not (0, default).
This step is very slow and MAY help to recover some TEs.
--anno [0|1] Perform (1) or not perform (0, default) whole-genome TE annotation after TE library construction.
--rmout [File] Provide your own homology-based TE annotation instead of using the EDTA library for masking. File is in RepeatMasker .out format. This file will be merged with the structural-based TE annotation. (--anno 1 required). Default: use the EDTA library for annotation.
--evaluate [0|1] Evaluate (1) classification consistency of the TE annotation. (--anno 1 required). Default: 0.
This step is slow and does not affect the annotation result.
--exclude [File] Exclude bed format regions from TE annotation. Default: undef. (--anno 1 required).
--threads|-t [int] Number of theads to run this script (default: 4)
--help|-h Display this help info
Identify intact elements of a paticular TE type:
1.Get raw TEs from a genome (specify -type ltr|tir|helitron in different runs)
perl EDTA_raw.pl [options]
--genome [File] The genome FASTA
--species [Rice|Maize|others] Specify the species for identification of TIR candidates. Default: others
--type [ltr|tir|helitron|all] Specify which type of raw TE candidates you want to get. Default: all
--overwrite [0|1] If previous results are found, decide to overwrite (1, rerun) or not (0, default).
--threads|-t [int] Number of theads to run this script
--help|-h Display this help info
2.Finish the rest of the EDTA analysis (specify -overwrite 0 and it will automatically pick up existing results in the work folder)
perl EDTA.pl --overwrite 0 [options]
If you developed a new TE method/got a TE library and want to compare it's annotation performance to the methods we have tested, you can:
1.annotate the rice genome with your test library:
RepeatMasker -e ncbi -pa 36 -q -no_is -norna -nolow -div 40 -lib custom.TE.lib.fasta -cutoff 225 rice_genome.fasta
2.Test the annotation performance of a particular TE category.
perl lib-test.pl -genome genome.fasta -std genome.stdlib.RM.out -tst genome.testlib.RM.out -cat [options]
-genome [file] FASTA format genome sequence
-std [file] RepeatMasker .out file of the standard library
-tst [file] RepeatMasker .out file of the test library
-cat [string] Testing TE category. Use one of LTR|nonLTR|LINE|SINE|TIR|MITE|Helitron|Total|Classified
-N [0|1] Include Ns in total length of the genome. Defaule: 0 (not include Ns).
-unknown [0|1] Include unknown annotations to the testing category. This should be used when
the test library has no classification and you assume they all belong to the
target category specified by -cat. Default: 0 (not include unknowns)
eg.
perl lib-test.pl -genome rice_genome.fasta -std ./EDTA/database/Rice_MSU7.fasta.std6.9.5.out -tst rice_genome.fasta.test.out -cat LTR
Please cite our paper if you find EDTA is useful:
Ou S., Su W., Liao Y., Chougule K., Agda J. R. A., Hellinga A. J., Lugo C. S. B., Elliott T. A., Ware D., Peterson T., Jiang N.✉, Hirsch C. N.✉ and Hufford M. B.✉ (2019). Benchmarking Transposable Element Annotation Methods for Creation of a Streamlined, Comprehensive Pipeline. Genome Biol. 20(1): 275.
Please also cite the software packages that were used in EDTA, listed in the EDTA/bin directory.
You may download the rice genome here.
If you have any issues with installation and usage, please check if similar issues have been reported in Issues or open a new issue. If you are (looking for) happy users, please read or write successful cases here.
I want to thank Jacques Dainat for contribution of the EDTA conda recipe as well as improving the codes. I also want to thank Qiushi Li, Zhigui Bao, Philipp Bayer, Nick Carleson, @aderzelle, Shanzhen Liu, Zhougeng Xu, Shun Wang, Nancy Manchanda, Eric Burgueño, and many more others for testing, debugging, and improving the EDTA pipeline.