alignment-nf
Nextflow pipeline for BAM realignment or fastq alignment



Description
Nextflow pipeline to perform BAM realignment or fastq alignment and QC, with/without local indel realignment and base quality score recalibration.
Dependencies
- Nextflow : for common installation procedures see the IARC-nf repository.
Basic fastq alignment
- bwa2 (default) or bwa
- samblaster
- sambamba
BAM files realignment
Adapter sequence trimming
ALT contigs handling
- the k8 javascript execution shell (e.g., available in the bwakit archive); must be in the PATH
- javascript bwa-postalt.js and the additional fasta reference .alt file from bwakit must be in the same directory as the reference genome file.
QC
Base quality score recalibration
- GATK4; wrapper 'gatk' must be in the path
- GATK bundle VCF files with lists of indels and SNVs (recommended: Mills gold standard indels VCFs, dbsnp VCF), and corresponding tabix indexes (.tbi)
A conda receipe, and docker and singularity containers are available with all the tools needed to run the pipeline (see "Usage")
Input
| Type | Description |
|---|---|
| --input_folder | a folder with fastq files or bam files |
Parameters
-
Mandatory
| Name | Example value | Description |
|---|---|---|
| --ref | hg19.fasta | genome reference with its index files (.fai, .sa, .bwt, .ann, .amb, .pac, and .dict; in the same directory) |
-
Optional
| Name | Default value | Description |
|---|---|---|
| --input_file | null | Input file (comma-separated) with 4 columns: SM (sample name), RG (read group ID), pair1 (first fastq of the pair), and pair2 (second fastq of the pair). |
| --output_folder | . | Output folder for aligned BAMs |
| --cpu | 8 | number of CPUs |
| --cpu_BQSR | 2 | number of CPUs for GATK base quality score recalibration |
| --mem | 32 | memory |
| --mem_BQSR | 10 | memory for GATK base quality score recalibration |
| --RG | PL:ILLUMINA | sequencing information for aligned (for bwa) |
| --fastq_ext | fastq.gz | extension of fastq files |
| --suffix1 | _1 | suffix for second element of read files pair |
| --suffix2 | _2 | suffix for second element of read files pair |
| --bed | bed file with interval list | |
| --snp_vcf | dbsnp.vcf | path to SNP VCF from GATK bundle (default : dbsnp.vcf) |
| --indel_vcf | Mills_1000G_indels.vcf | path to indel VCF from GATK bundle (default : Mills_1000G_indels.vcf) |
| --postaltjs | bwa-postalt.js" | path to postalignment javascript bwa-postalt.js |
| --feature_file | null | Path to feature file for qualimap |
| --multiqc_config | null | config yaml file for multiqc |
| --adapterremoval_opt | null | Command line options for AdapterRemoval |
| --bwa_mem | bwa-mem2 mem | bwa-mem command; use "bwa mem" to switch to regular bwa-mem (both are in the docker and singularity containers) |
-
Flags
Flags are special parameters without value.
| Name | Description |
|---|---|
| --help | print usage and optional parameters |
| --trim | enable adapter sequence trimming |
| --recalibration | perform quality score recalibration (GATK) |
| --alt | enable alternative contig handling (for reference genome hg38) |
| --bwa_option_M | Trigger the -M option in bwa and the corresponding compatibility option in samblaster (marks shorter split hits as secondary) |
Usage
To run the pipeline on a series of fastq or BAM files in folder input and a fasta reference file hg19.fasta, one can type:
nextflow run iarcbioinfo/alignment-nf -r v1.3 -profile singularity --input_folder input/ --ref hg19.fasta --output_folder outputTo run the pipeline without singularity just remove "-profile singularity". Alternatively, one can run the pipeline using a docker container (-profile docker) the conda receipe containing all required dependencies (-profile conda).
Use bwa-mem instead of bwa-mem2
To use bwa-mem, one can type:
nextflow run iarcbioinfo/alignment-nf -r v1.3 -profile singularity --input_folder input/ --ref hg19.fasta --output_folder output --bwa_mem "bwa mem"Enable adapter trimming
To use the adapter trimming step, you must add the --trim option, as well as satisfy the requirements above mentionned. For example:
nextflow run iarcbioinfo/alignment-nf -r v1.3 -profile singularity --input_folder input/ --ref hg19.fasta --output_folder output --trimEnable ALT mode
To use the alternative contigs handling mode, you must provide the path to an ALT aware genome reference (e.g., hg38) AND add the --alt option, as well as satisfy the above-mentionned requirements. For example:
nextflow run iarcbioinfo/alignment-nf -r v1.3 -profile singularity --input_folder input/ --ref hg19.fasta --output_folder output --postaltjs /user/bin/bwa-0.7.15/bwakit/bwa-postalt.js --altEnable base quality score recalibration
To use the base quality score recalibration step, you must provide the path to 2 GATK bundle VCF files with lists of known snps and indels, respectively, AND add the --recalibration option, as well as satisfy the requirements above mentionned. For example:
nextflow run iarcbioinfo/alignment-nf -r v1.3 -profile singularity --input_folder input/ --ref hg19.fasta --output_folder output --snp_vcf GATKbundle/dbsnp.vcf.gz --indel_vcf GATKbundle/Mills_1000G_indels.vcf.gz --recalibrationOutput
| Type | Description |
|---|---|
| BAM/ | folder with BAM and BAI files of alignments or realignments |
| QC/BAM/multiqc_qualimap_flagstat_*report.html | multiQC report for qualimap and samtools flagstat (duplicates) |
| QC/BAM/multiqc_qualimap_flagstat_*report_data | data used for the multiQC report |
| QC/qualimap/file_BQSRecalibrated.stats.txt | qualimap summary file |
| QC/qualimap/file_BQSRecalibrated/ | qualimap files |
| QC/BAM/BQSR/ | GATK base quality score recalibration outputs (tables and pdf comparing scores before/after recalibration) |
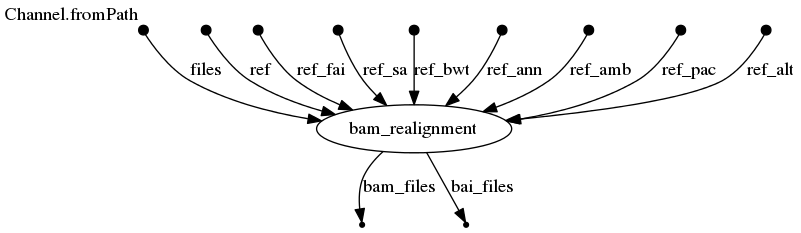
Directed Acyclic Graph
FAQ
Why did Indel realignment disappear from version 1.0?
Indel realignment was removed following new GATK best practices for pre-processing.
Contributions
| Name | Description | |
|---|---|---|
| Nicolas Alcala* | AlcalaN@fellows.iarc.fr | Developer to contact for support |
| Catherine Voegele | VoegeleC@iarc.fr | Tester |
| Vincent Cahais | CahaisV@iarc.fr | Tester |
| Alexis Robitaille | RobitailleA@students.iarc.fr | Tester |