This dataset contains AlphaFold's predictions and internal latent representations for a large subset* of the proteins that are referenced in Papyrus (a large scale curated dataset aimed at bioactivity predictions).
*Note: At the current date (17/06/2022) - 3626/6000+ proteins contained in Papyrus dataset are fully processed.
Why is this dataset useful?
- Even though an extensive database of AlphaFold’s predictions of protein 3D structures is already publicly available, the features that the model uses internally have not yet been published. Since the final output of AlphaFold is the set of 3D coordinates of the protein backbone, one could argue that this format is highly condensed (given the huge scale-down in dimensionality). It is likely that there is a lot more valuable information that can be captured within this large transformer model.
- Additionally, since processing multiple proteins through this model takes a considerable amount of time and compute, doing it multiple times is somewhat wasteful. After processing a protein sequence the generated embeddings and predictions can be saved and reused for other projects.
Definitions for each internal representation of proteins used within AlphaFold can be found in the supplementary material of the original publication. Below, each representation will be referred to using the same notation as in the paper.
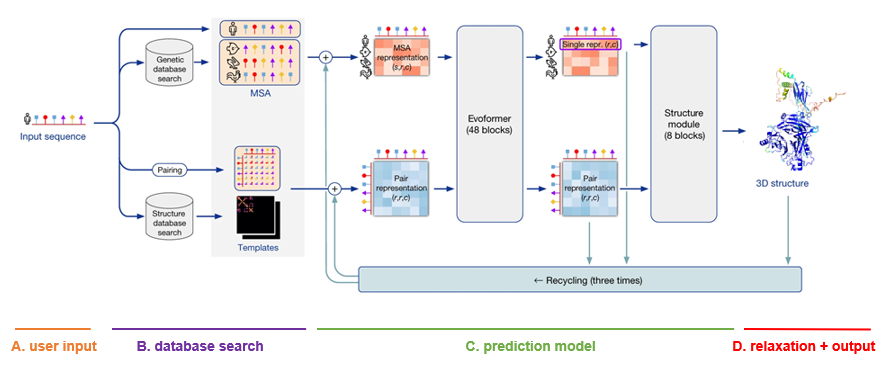
- MSA module queries protein databases to find similar amino acid sequences, performs multiple sequence alignment and creates the initial MSA representation
$(m_{si})$ . This contains all the relevant information about evolutionarily related protein sequences. -
Pair representation
$(z_{ij})$ is created by querying protein structure databases, trying to find similar proteins with known structure and then building feature vectors for each residue pair based on this information.
- Evoformer - a very large transformer that iteratively cross-integrates information from the MSA representation
$(m_{si})$ and pair representation$(z_{ij})$ using a wide variety of attention mechanisms. One of its outputs is the single representation$(s_i)$ which condenses all the MSA information into a single vector (per residue). - Structure module - a point invariant transformer that uses features generated by the evoformer to iteratively modify a 3D graph of the protein backbone and produce the final prediction. After the last layer of this transformer another structure representation
$(a_i)$ can be exctracted.
- Post processing step that ensures that there are no violations and clashes in the generated 3D structure and attempts to fix it if needed.
Two versions of the dataset are available:
- Lightweight - contents listed in the table below. This is the default option when downloading (1-10MB per protein).
-
Full - includes MSA representation
$(m_{si})$ , pair representation$(z_{ij})$ and distogram logits. Drastically increases the size of the dataset (+~100-1000MB per protein). Currently full data is only available for a small subset of proteins.
↓ data/* -> main data directory
↓ data/PID/* -> data of a single protein of length L
Filename Description Tensor shape Lightweight single.npy* ( $s_i$ ) evoformer single representation[L x 384] ✔️ structure.npy* $(a_i)$ output of the last layer of structure module[L x 384] ✔️ msa.npy* $(m_{si})$ processed MSA representation[N x L x 256] pair.npy* $(z_{ij}$ ) evoformer pair representation[L x L x 128] PID.pdb 3D protein structure prediction ✔️ PID_unrelaxed.pdb 3D protein structure prediction w/o relaxation step (D) ✔️ confidence.npy* confidence in structure prediction (0-100) 1 ✔️ plldt.npy* confidence in structure prediction per residue [L] ✔️ PID.fasta protein amino acid sequence and metadata ✔️ timings.json Processing log ✔️
↓ data/PID2/* -> data of protein #2
...
*Note: L: sequence length, N: number of aligned sequences via MSA.
First clone the repository:
git clone https://github.com/andriusbern/foldedPapyrus && cd foldedPapyrus-
Single proteins
Data of individual proteins can be downloaded using their accession numbers.
python download.py --pid P14324
-
Partial dataset
You can use a file containing a list of protein accession numbers separated by a newline “\n” and pass it via command line. You can find examples of these protein subset files in the subsets/ directory.
python download.py --pid_file subsets/kinases.txt
-
Full dataset
Alternatively if you want to download all the data that is currently available:
python download.py --all
- If you are trying to download a lot of data you might get a "HTTP Error 429: Too many requests" error. In that case you should wait an hour or so before downloading again.
- AlphaFold actually consists of 5 separate models with slightly different architectures and hyperparameters. The normal pipeline of processing a single protein involves all 5 of these models whose predictions are then ranked based on the confidence metrics. Since this requires 5x the time/compute, this dataset was created by only running the [Model 0]. In the future this dataset could be expanded to include predictions from all models (if there is a need for it).
- This dataset was produced by modifying AlphaFold code, parallelising it and optimising the throughput of CPU and GPU intensive parts of the code. By now the pipeline is very automated so if you need embeddings for specific proteins, please contact me at a.bernatavicius@liacs.leidenuniv.nl, I will be glad to help.
- Béquignon OJM, Bongers BJ, Jespers W, IJzerman AP, van de Water B, van Westen GJP. Papyrus - A large scale curated dataset aimed at bioactivity predictions. ChemRxiv. Cambridge: Cambridge Open Engage; 2021; This content is a preprint and has not been peer-reviewed.
- Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). https://doi.org/10.1038/s41586-021-03819-2